Intro Genome Bio 2013: Difference between revisions
Jump to navigation
Jump to search
BIOL 105. Introduction to Genome Biology (Fall 2013)
Instructor: Dr Weigang Qiu, Associate Professor of Biology
Room: C104 Hunter North (a Computer Lab)
Hours: Tuesdays, 12:45-3:15PM
Office Hours: Room 839 HN; Tuesday 5-7pm or by appointment. Phone: 212-772-5296.
Email: weigang@genectr.hunter.cuny.edu (I will advise students before and after the class as well as during office hours. Emails will be answered only in special situations.)
Volunteer Tutors: Slav Kendall<sviatoslav.kendall at gmail.com> & Rayees Rahman<rayees.rahman at live.com>
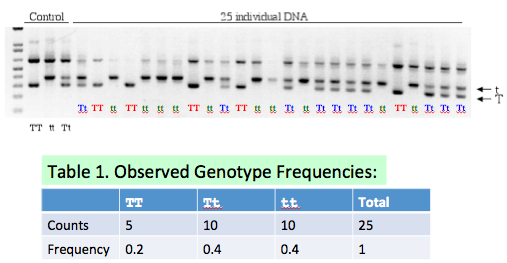
Table 1 Observed Counts:
Table 2. Expected Counts (if there is no association between the genotypes and the disease phenotype)
imported>Weigang |
imported>Weigang |
||
| (155 intermediate revisions by 2 users not shown) | |||
| Line 3: | Line 3: | ||
<center>'''Room:''' C104 Hunter North (a Computer Lab)</center> | <center>'''Room:''' C104 Hunter North (a Computer Lab)</center> | ||
<center>'''Hours:''' Tuesdays, 12:45-3:15PM</center> | <center>'''Hours:''' Tuesdays, 12:45-3:15PM</center> | ||
<center>'''Office Hours:''' Room 839 HN; | <center>'''Office Hours:''' Room 839 HN; Tuesday 5-7pm or by appointment. '''Phone:''' 212-772-5296.</center> | ||
<center>''' | <center>'''Email''': weigang@genectr.hunter.cuny.edu (I will advise students before and after the class as well as during office hours. <u>Emails will be answered only in special situations.</u>) </center> | ||
<center><font color="red">Volunteer Tutors</font>: Slav Kendall<sviatoslav.kendall at gmail.com> & Rayees Rahman<rayees.rahman at live.com></center> | |||
---- | ---- | ||
==General Information== | ==General Information== | ||
[[File:BIOL105-title.jpg|thumbnail|center|Content Diagram]] | |||
===Course Description=== | ===Course Description=== | ||
* '''Background | * '''Background''': A genome is the total genetic content of an organism. Driven by breakthroughs such as the decoding of the first human genome and rapid DNA-sequencing technologies, biomedical sciences are undergoing a rapid and profound transformation into a highly data-intensive field, which requires familiarity with concepts in both biology and computer science. Genome information is revolutionizing virtually all aspects of biology and medicine and will lead to major advances such as more efficient production of renewable energy, better cures for cancers, and longer and healthier life expectancy. | ||
* '''Contents | * '''Contents''': This course will introduce genome-sequencing technologies, explore genome projects online, and discuss both the benefits and challenges (e.g., ethical and legal) of the genomic revolution to society. Each session will consist of lecture instructions and computer-based laboratory practices. | ||
* ''' | * '''Learning Goals''' | ||
* ''' | ** '''Human Genome''': Knowing the basic content and structure of the human genome | ||
* | ** '''Central Dogma''': Understanding the Central Dogma and how genes are regulated (i.e., turned on/off) | ||
** | ** '''Genome diversity''': Knowing the three major divisions of life, the use of DNA/protein sequences to classify organisms, and mechanisms of genome evolution | ||
** | ** '''Bioinformatics''': Understanding the importance of computation and statistics in producing, analyzing, and disseminating genome information | ||
** | * '''Learning Outcomes''' | ||
** | ** Be able to describe the structure and function of the human genome | ||
* ''' | ** Be able to find chromosomal location, function, disease-association, and variability of a human gene using scientific databases | ||
** Be able to identify and classify organisms based on their DNA sequences | |||
** Be able to describe the processes of genome evolution, including mutation, duplication, recombination, horizontal gene transfer, genetic drift, and natural selection | |||
* '''Textbook:''' Arthur Lesk (2012). ''Introduction to Genomics'' (2nd Edition). Oxford University Press. [http://www.amazon.com/Introduction-Genomics-Arthur-M-Lesk/dp/0199564353/ref=sr_1_1?ie=UTF8&qid=1378143035&sr=8-1&keywords=introduction+to+genomics Amazon Link] | |||
* '''Academic Honesty:''' Hunter College regards acts of academic dishonesty (e.g., plagiarism, cheating on examinations, obtaining unfair advantage, and falsification of records and official documents) as serious offenses against the values of intellectual honesty. The College is committed to enforcing the CUNY Policy on Academic Integrity and will pursue cases of academic dishonesty according to the Hunter College Academic Integrity Procedures. | * '''Academic Honesty:''' Hunter College regards acts of academic dishonesty (e.g., plagiarism, cheating on examinations, obtaining unfair advantage, and falsification of records and official documents) as serious offenses against the values of intellectual honesty. The College is committed to enforcing the CUNY Policy on Academic Integrity and will pursue cases of academic dishonesty according to the Hunter College Academic Integrity Procedures. | ||
===Grading Policy=== | ===Grading Policy=== | ||
* '''Treat assignments as take-home exams.''' Student performance will be evaluated by weekly assignments and | * '''Treat assignments as take-home exams.''' Student performance will be evaluated by weekly assignments and three exams. While students are allowed to work in groups for assignments (and answers to some questions are available on the textbook website), students are expected to compose the final answers independently. Writings that are virtually exact copies to each other or to the web answers will be considered plagiarism. Zero points will be given to ALL involved individuals if the instructor considers there is enough evidence for plagiarism. To avoid being investigated for plagiarism, <u>Do NOT copy or let others copy your work</u>. | ||
* '''Submit assignments in Printed Hard Copies.''' Email attachments will NOT be accepted. Each assignment will be graded based on timeliness ( | * '''Submit assignments in Printed Hard Copies.''' Email attachments will NOT be accepted. Each assignment will be graded based on timeliness (20%), effort (40%), and correctness (40%). | ||
* ''' | * Since each session will consist of a web-based practical session, '''attendance''' is required and part of the grade. | ||
** Assignments | * '''Composition of your final grade''' | ||
** Mid-term | ** Assignments: weekly assignments, totaling 100 pts | ||
** Mid-term and Final Comprehensive Exams: 3 mid-terms X 50 points each = 150 pts | |||
** | ** Attendance and active classroom participation (50 pts): 1 unexcused absences = 40 pts; 2 absences = 30pts; More than 2 = 0. Points will be deducted for lack of participation in computer-lab exercises. | ||
=== | ==Weekly Schedule (All Tuesdays)== | ||
===September 3. Introduction to Genomics (1)=== | |||
# Lecture Slides: [[File:BIOL105-Intro-Genomics.pptx|thumbnail]] | |||
# Course Overview | |||
# Contents of the human genome (Chapter 1, pg. 3-10) | |||
# Genome organization (Chapter 1, pg. 10-17) | |||
# Assignment 1 (15 pts, due 9/10) [Finalized on 9/5, 7pm] | |||
## (2 pts) '''(Note: modified from the textbook)''' Exercise 1.1. (a) Make a very rough estimate of the average density (e.g., number of genes per million base pairs) of protein-coding genes in the human genome, assuming total genome size ~ 3 x 10^6 bp and total number of genes ~ 3 X 10^4 genes. <pre style="white-space: pre-wrap;">Answer: 3x10^4 (genes)/3x10^9 (bp; typo in the original question)=1 gene every 10,000 bp</pre>(b) Further assuming that the average gene length is 1000 bp, what is the proportion of bases that code for proteins? <pre style="white-space: pre-wrap;">Answer: 3x10^4x10,000 (bp in genes)/3x10^9 (bp, total)=1x10^-2, or 1% of the total bases code for genes</pre> | |||
## (4 pts) Exercise 1.3. For the standard genetic code (see lecture slides), give an example of a pair of codons related by a synonymous single-site substitution (a) at the third position and (b) at the first position. (c) Give an example of a pair of codons related by a non-synonymous single-site substitution at the third position. (d) Can a change in the second position of a codon ever produce a synonymous mutation? <pre style="white-space: pre-wrap;">Answer to (d): No. All 2nd position changes are non-synonymous. Stop codons are not "synonymous" amino acids.</pre> | |||
## (4 pts) Exercise 1.4. (a) Is it possible to convert Phe to Tyr by a single base change? If so, what would be possible wild-type and mutant codons? <pre style="white-space: pre-wrap;">Answer: Yes. Either TTT (Wild Type, WT) to TAT (Mutant, MU), or TTC (WT) to TAC (MU). Both are changes at the 2nd codon position.</pre>(b) Is it possible to convert Ser to Arg by a single base change? If so, what would be possible wild-type and mutant codons? <pre style="white-space: pre-wrap;">Answer: Yes. Either AGT (Wild Type, WT) to CGT (Mutant, MU), or AGC (WT) to CGC (MU). Both are changes at the 1st codon position.</pre>(c) What is the minimum number of base substitutions that would convert Cys to Glu? <pre style="white-space: pre-wrap;">Answer: Three. Cys codons are either TGT or TGC, and Glu codons are either GAA or GAG. These two amino acids don't share the first, second, or the third codon positions.</pre>(d) In the evolution off an essential protein encoded by a single gene, a Trp is converted to a Gln by two successive single-base changes. What is the intermediate codon?<pre style="white-space: pre-wrap;">Answer: TGG (Trp) through CGG (Arg, 1st position change) to CAG (Gln, 2nd position change). The other possibility is highly unlikely since it goes through a stop codon: TGG through TAG (Stop, 2nd position change) to CAG (Gln, 1st position change)</pre> | |||
## (5 pts) Weblem: Use the NCBI Human Genome Resource webpage to identify diseases associated with heamoglobin alpha-1 (HBA1). Summarize its association with the disease "alpha-thalassemia". Include key references and a discussion on why this is a case against "genetic determinism". <pre style="white-space: pre-wrap;">Answer: [http://www.omim.org/entry/141800 Online Mendelian Inheritance in Man database Entry 141800] lists 5 diseases associated with HBA1 mutations. See [http://www.omim.org/entry/604131 See OMIM Record #604131] for information on alpha-thalassemia. The key reference is Weatherall (2001), which concludes that heritability of alpha-thalassemia is low and environmental factors influences the disease outcome. Mutations at HBA1 locus alone is not a sufficient predictor of the disease, an argument against "genetic determinism" (At the least, it's not a single-gene genetic disease).</pre> | |||
===September 10. Introduction to Genomics (2)=== | |||
# Lecture Slides: [[File:Session-2-intro-to-genomics-part-2.pptx|thumbnail]] | |||
# Assignment 2 (Finalized on 9/11 @11pm) | |||
## (2 pts) (Modified from Exercise 1.8). Using the NCBI human genome browser, find out which of the following bands of human chromosome 16 are gene-rich: p13.3, q22.1, or q11.2? <pre style="white-space: pre-wrap;">Answer: According to the NCBI Human Genome Resource page (at the 1/100 zoom level and using "recenter"), the q22.1 has 32 genes, q11.2 has 0 genes, and p13.3 has 74 genes. So q13.3 and q22.1 (both light-colored) are gene-rich, while q11.2 (darker-colored) is gene-poor.</pre> | |||
## (4 pts) (Modified from Exercise 1.11). Make a printout of the GenBank file with the Accession "V00488" (print ONLY the part with the DNA sequence). Mark the following sequence regions with highlighters: (a) all introns, (b) all exons, (c) 5'-UTR and 3'-UTR, (d) start and stop codons. (Hint: Use the "Graphics" view with zooming.) <pre style="white-space: pre-wrap;">Answer: Using the Graphics view and zoom in at the nucleotide level, you will find the following coordinates: Primary transcript: 98-929, Exon 1: 98-230 (most likely 98-229), Exon 2: 348-551 (should be 347-551), Exon 3: 692-929, Intron 1: 231-347 (should be 230-346, starting with GT and ending in AG), Intron 2: 552-691, 5, Start Codon: 135-137, Stop Codon: 818-820, 5'-UTR: 98-134, 3'-UTR: 821-929</pre> | |||
## (4 pts) (Modified from Exercise 1.12). On a separate printout of the GenBank file with the Accession "V00488" (print ONLY the part with the ''amino-acid'' sequence). Mark the following regions with highlighters: (a) amino acids coded for by exon 1, (b) by exon 2, (c) by exon 3. (Hint: Use the "Graphics" view with zooming). (D) Is it possible that a single codon is split by an intron (e.g., 1st codon position on exon 1 and 2nd & 3rd nucleotides on exon 2) ? | |||
<gallery align="center"> | |||
File:Q3-answerkey.png|Answer key for 1.11 | |||
File:Q4-answerkey.png|Answer key for 1.12 | |||
</gallery> | |||
===September 17. Introduction to Genomics (3)=== | |||
# Lecture Slides: [[File:Session-3.pptx|thumbnail]] | |||
# Genome database (Chapter 1, pg.24-30) | |||
# Cancer genomics: [http://cancergenome.nih.gov/newsevents/multimedialibrary/videos/thegeneticbasisofcancer The Cancer Genome Atlas (TCGA) Project] | |||
# Genome evolution (Chapter 1. pg.30-35) | |||
# Assignment 3 (Finalized on 9/18, Thursday, 11:55pm) | |||
## (6 pts). Problem 1.6 (on page 38). Instructor's Note: (1) Mark your answers on a printout or copy of Figure 1.17(a); (2) for part (d), show your work. A simple choice without reasoning or explanation will NOT get full credits. | |||
## (4 pts). Read/Watch the following links and extract one example to describe how genome sequencing helps health care. | |||
### [http://hapmap.ncbi.nlm.nih.gov/healthbenefit.html HapMap benefits] | |||
### [http://cancergenome.nih.gov/cancergenomics/impact Impact of cancer genomics] | |||
### [http://cancergenome.nih.gov/newsevents/multimedialibrary/videos/personalizedcancertherapies Link between cancer genomics and personalized therapy] | |||
===September 24. First Mid-term Exam=== | |||
* Review session: 12:45-1:40 | |||
* Exam Part I. 1:45-2:30. No computer access | |||
* Exam Part II. 2:30-3:15. Computer-based Practicum (NCBI websites only) | |||
===October 1. Genome Structure and Function (1)=== | |||
# Lecture Slides: [[File:Session-4.pptx|thumbnail]] (updated) | |||
# Classic Genetics (Chapter 3, pg.80-82) | |||
# Structure of DNA (Chapter 3, pg. 90-93) | |||
# DNA replication and PCR (Chapter 3, pg 93-95) | |||
# Assignment 4 (Finalized as of 10/2, 1pm) | |||
## (5 pts). Two alleles (T and t) are segregating at a single genetic locus In a population of diploid individuals. "T" is the dominant allele and "t" is recessive for a phenotype (e.g., "Taster" and "Non-taster"). If two parents are both heterozygotes at this locus, what is the expected Mendelian ratio of genotypes of their offspring? Mendelian ratio of the phenotypes? Show your work.<pre>Genotypes: TT:Tt:tt=1:2:1; Phenotypes: Taster:Non-taster=3:1</pre> | |||
## (5 pts) Answer the following questions about the Polymerase Chain Reaction (PCR): | |||
### Describe the three steps in each round of PCR, including the approximate temperature and the reaction occurring at each step <pre>1. Denature DNA into single strands (95ºC); 2. Anneal primers (50ºC); 3. Synthesize DNA (72ºC)</pre> | |||
### What are the key chemical ingredients required for amplifying a DNA region using PCR? <pre>DNA template, primers, DNA polymerase, and nucleotides</pre> | |||
### Starting from a single DNA molecule (note: double-stranded), what is the total number of copies of a gene on this DNA after 20 rounds of PCR amplification? Show your work. <pre>2^20=1,048,576</pre> | |||
## (5 pts) Fill in Table 1 on Slide #12 based on the gel picture. Explain the basis of your counts and frequencies. | |||
[[File:HW4 Q3.jpg|center|framed]] | |||
===October 8. Genome Structure and Function (2)=== | |||
# Lecture Slides: [[File:Session-5.pptx|thumbnail]] (updated) | |||
# Test of PTC genotype-phenotype association | |||
# Linkage Map (Chapter 3, pg.82-85) | |||
# Automated Sequencing (Chapter 3, pg 97-104) | |||
# Assignment 5.(Finalized on Friday, 10/11/2013, 12 noon; Updated o nTuesday 10/15/2013) | |||
## (2 pts) Exercise 3.2 (pg.110). Explain.<pre style="white-space: pre-wrap;">D=A1B1 x A2B2 - A1B2 x A2B1=0.2x0.45-0.15x0.2= 0.06. D is a measure of linkage disequilibrium (LD) between two loci. When D=0, it means that there is no association between allele frequencies on two loci, or no LD. In this case, D>0, suggesting that chromosome with A1B1 and A2B2 are over-represented (with respect to expectation) in the population.</pre> | |||
## (3 pts) Exercise 3.6 (pg.110). Explain.<pre style="white-space: pre-wrap;">The bottom band in the middle lane corresponds to the 4 kb BamHI fragment. The 2nd band from the top in the right lane corresponds to the smallest (1.5 kb, with lowest molecular weight) EcoRI fragment. In a gel electrophoresis, DNA molecules travel from the negative electrode towards the positive electrode. This is because DNA is negatively charged in neutral conditions. Smaller DNA fragments, which have lower molecular weights, travel faster than longer ones. Note: 1 kb = 1 kilobases = 1,000 base pairs</pre> | |||
## (5 pts) Problem 3.6 (pg.113). Explain. Hints: See figure at right for an illustration of the RNA stem and loop [[File:Rna-loop.jpg|thumbnail|RNA structure]]. Compare your results with those produced by this web-based RNA structure predictor: [http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi RNAfold web server] <pre style="white-space: pre-wrap;">(a) DNA synthesis occurs in the direction of 5' to 3', so the shortest synthesized fragment is at the bottom. Therefore, the sequence is: 5'-CCTTATCTCTTGATAGGGAACCGGTTTCCCGGC-3'. (b) There is no long (6 bp or more) hairpin structure predicted for this sequence. The longest exact self-complementary region predicted by the RNAfold program is a 4-bp stem near the 3' end: 5'-CCTTATCTCTTGATAGGGAA{CCGG}TTTC{CCGG}C-3'. (c). Based on the previous analysis, this region of DNA is more likely to encode protein.</pre> | |||
## (5 pts) Perform a hypothetical genotype-phenotype association study. Individuals are categorized depending on having a disease ("Case") or not ("Control"). Each individual is genotyped at one SNP site with two possible states (e.g., "A" or "G"). The following table is a 2X3 contingency table from the study. Perform a chi-squared test using [http://www.physics.csbsju.edu/stats/contingency_NROW_NCOLUMN_form.html this website]. Answer the following questions: | |||
### What are the expected counts if there is no association between the genotype and the disease phenotype? (Null hypothesis) <pre>See Table 2 below</pre> | |||
### What is your chi-square value and the p value? What is your interpretation of the p value? Using a cutoff of p=0.05, what can you conclude from the study (i.e., does it show a statistically significant association between genotype and disease?) <pre style="white-space: pre-wrap;">chi-square = 8.97, probability (p value) = 0.011, which is the probability of obtaining observed counts (including more biased counts than these) by chance (i.e., if there is no association between the genotypes and the disease). If p<0.05 is considered statistically significant, the result suggest that the null hypothesis ("no association") could be rejected and there is a statistically significant association between the genotype and the disease.</pre> | |||
### According to your comparisons between observed and expected counts, which genotype is the most over-represented in individuals with diseases? Which genotype is the most under-represented with diseases? <pre style="white-space: pre-wrap;">Compare the observed counts (in Table 1) with corresponding expected counts (Table 2). The "GG" genotype is the most over-represented and the "AA" genotype is the most under-represented in the disease cases.</pre> | |||
<center> Table 1 Observed Counts: | |||
{| class="wikitable" | |||
|- | |||
! !! AA !! AG !! GG | |||
|- | |||
| Case || 221 || 198 || 55 | |||
|- | |||
| Control || 279 || 212 || 35 | |||
|} | |||
</center> | |||
<center> Table 2. Expected Counts (if there is no association between the genotypes and the disease phenotype) | |||
{| class="wikitable" | |||
|- | |||
! !! AA !! AG !! GG | |||
|- | |||
| Case || 237 || 194 || 43 | |||
|- | |||
| Control || 263 || 216 || 47 | |||
|} | |||
</center> | |||
===October 15. No Class (Monday Schedule)=== | ===October 15. No Class (Monday Schedule)=== | ||
===October 22. | ===October 22. Genome Structure and Function (3)=== | ||
===October 29=== | * Lecture Slides: [[File:BIOL105-Session-6.pptx|thumbnail]] | ||
===November 5=== | * [http://learn.genetics.utah.edu/content/labs/microarray/ Virtual Lab: Microarray] | ||
===November 12=== | * Videos for understanding genome functions: | ||
===November 19=== | {| class="wikitable" | ||
===November 26=== | |- | ||
===December 10=== | ! Transcription !! Splicing !! Translation | ||
===December 17. Final Exam=== | |- | ||
| {{#ev:youtube|WsofH466lqk|300|center|Transcription}} || {{#ev:youtube|FVuAwBGw_pQ|300|center|Splicing}} || {{#ev:youtube|5bLEDd-PSTQ|300|center|Translation}} | |||
|} | |||
* Assignment 6 (10 pts). Use [http://genome-www.stanford.edu/nci60/search.shtml the Cancer Transcriptome Website] for the following exercises: | |||
** Access the expression data for protein kinase C Beta (PRKCB1). | |||
** Name a group of cancers associated with underexpression of PRKCB1. Attach the figure and show your rationale. <pre style="white-space: pre-wrap;">Green color indicates under-expression of a gene in cancer cells (relative to control, healthy cells). Figure 1 (see below) shows that PRKCB1 is under-expressed in, e.g., colon and breast cancer cells.</pre><font color="red">Notice: Flipped answers to this and the next question are acceptable since the color scheme is not clearly specified on the website.</font> | |||
** Name a group of cancers associated with overexpression of PRKCB1. Attach the figure and show your rationale. <pre style="white-space: pre-wrap;">Red color indicates over-expression of a gene in cancer cells (relative to control, healthy cells). Figure 1 (see below) shows that PRKCB1 is over-expressed in leukemia cells.</pre> | |||
** Name 3 genes with similar cancer expression patterns as PRKCB1. Attach the figure and show your rationale. <pre style="white-space: pre-wrap;">Figure 2 (see below) shows that PLCE, CD53, PTPRC, CDW52, LRMP, RHAG, and PDCD genes have similar expression patterns among cancer cells as PRKCB1.</pre> | |||
[[File:Cancer-array-1.png|400px|thumbnail|center|Figure 1]] | |||
[[File:Cancer-array-2.png|400px|thumbnail|center|Figure 2]] | |||
===October 29. Genome Structure and Function (4)=== | |||
* Lecture Slides: [[File:Session-7.pptx|thumbnail]] | |||
* Cancer Genomics: Field trip to [http://ccsbsymposium.mskcc.org/Agenda.aspx 3rd Annual Symposium of Systems Biology of Cancer at MSKCC]. We will attend the '''Afternoon Session B''', which has three talks during 1:30-3pm. Take notes and hand in one copy | |||
* Assignment #7. (Finalized Wed, 10/30, 3pm) | |||
# (2 pts) Exercise 3.14 (pg. 111). Show your work.<pre style="white-space: pre-wrap;">see "Contig 2" in the following figure</pre>[[File:Contig.jpg|thumbnail]] | |||
# (3 pts) Exercise 3.15 (pg. 111). Show your work. [Hint: Enlarge the picture (e.g., double the size), and then measure the lengths of individual fragments and the region with a ruler. Read the measurements in millimeters.] <pre style="white-space: pre-wrap;">(a) The minimum coverage of any position is zero (in the gap region, see figure at right), (b) the maximal coverage of any position is at the red vertical line (see figure at right), which is covered by 9 individual reads. (c) The average coverage of the entire region is the sum of lengths of all reads divided by the length of the region. The answer is approximately 4, indicating that on average each DNA position is covered by 4 sequencing reads. </pre> | |||
# (5 pts) Search the NCBI Gene database for "BRCA1" gene. Based on [http://www.ncbi.nlm.nih.gov/gene/672 this Gene Card for the human version of the gene], summarize its molecular function, chromosomal location, gene structure (number of exons and introns, lengths of primary transcript, mRNA, and protein), mutations associated with cancers. Follow the link to OMIM to find out what are the 185delAG and 5382insC mutations (base substitution, deletion, insertion, consequence to protein synthesis?) and in what exons do they appear. | |||
## '''Molecular function''': "This gene encodes a nuclear phosphoprotein that plays a role in maintaining genomic stability, and it also acts as a tumor suppressor...This protein thus plays a role in transcription, DNA repair of double-stranded breaks, and recombination." | |||
## '''Chromosomal location''': Chromosome 17: 17q21 | |||
## '''Gene structure''': Five splicing variants (or isoforms) of BRCA1 are recorded in [http://www.ncbi.nlm.nih.gov/nuccore/NC_000017.10?report=genbank&from=41184133&to=41289677&strand=true this GenBank file]. For example, Isoform 4 has a primary transcript from base #13,546 to 93,366 (a total of 93366-13546+1=79,821 bases), 21 introns, 22 exons, a protein sequence of 759 amino acids (see [http://www.ncbi.nlm.nih.gov/protein/237681123 protein_id="NP_009229.2"]), an mRNA with 3 times protein length, plus 5'- and 3'-UTRs (approximately 3 x 759=2,277 bases). | |||
## '''Mutations associated with cancers''': "Mutations in this gene are responsible for approximately 40% of inherited breast cancers and more than 80% of inherited breast and ovarian cancers. " See two examples below. | |||
## These are deletion/insertion (indel) mutations in exons and thus cause frame shifts and lead to truncated proteins. The '''[http://omim.org/entry/113705#0003 185delAG mtuation]''' is a "2-bp (AG185) deletion in the normal sequence TTA GAG of codons 22-23 in exon 3. The AGAG presumably predisposed to the deletion. This mutation changes the reading frame of the mRNA and causes a premature termination codon at position 39." The '''[http://omim.org/entry/113705#0018 5382insC mutation]''' is "a 1-bp (C) insertion at position 5382 in exon 20, changing the reading frame of the mRNA and causing a premature termination codon at position 1829 in exon 24." | |||
[Hint: See following youtube video for different types of mutations: {{#ev:youtube|kp0esidDr-c|300|center|Frameshift mutations}} | |||
===November 5. 2nd Mid-term Exam=== | |||
# 12:45-1:45. Review | |||
# 1:45 - 2:30. Exam without computer access | |||
# 2:30 - 3:15. Computer-based practicum | |||
===November 12. Genome Evolution (1)=== | |||
* Lecture Slides: [[File:Session-8.pptx|thumbnail]] | |||
* Workshop I. Molecular population genetics and classification (Chapter 2) | |||
* (5 pts) Assignment 8 (Finalized on 11/14, 3pm). Using haplotype sequences in Table 2.2 (pg 75), compute sequence differences between each pair of Clade I, II, III, and IV. Within each clade, use the '''last''' sequence as the representative. Your answers should include the following items (and show your procedures, not just the answers): | |||
** Copy and paste of four chosen sequences | |||
** ''Counts'' of nucleotide differences between each pair of sequences (a total of 6 pairs) | |||
** ''Fraction'' of sequence differences for each pair, using the total length of 294 bases | |||
** Estimates of ''divergence time'' (in years) between each pair of sequences, assuming a sequence-divergence rate of 0.125 changes per 10,000 years. | |||
Hint: Present your answers using tables like this one: | |||
{| class="wikitable" | |||
|- | |||
! !! Clade I !! Clade II !! Clade III !! Clade IV | |||
|- | |||
| Clade I || 0 || 11 base differences; 11/294=0.0374 diff per base; 0.0374/0.125 x 10,000=2993 years of divergence|| 11;0.0374; 2993 || 20; 0.0680; 5442 | |||
|- | |||
| Clade II || (blank) || 0 || 18; 0.0612; 4898 || 23; 0.0782; 6259 | |||
|- | |||
| Clade III || (blank) || (blank) || 0 || 23; 0.0782; 6259 | |||
|- | |||
| Clade IV || (blank) || (blank) || (blank) || 0 | |||
|} | |||
===November 19. Genome Evolution (2)=== | |||
* Lecture Slides: [[File:Session-9.pptx|thumbnail]] | |||
* Workshop I. Molecular population genetics and classification (Chapter 2) | |||
* Workshop II. Origin of Native Americans | |||
** [http://www.sciencemag.org/content/342/6157/409.full Report in the Science Journal] | |||
** [http://www.sciencemag.org/content/342/6157/409/suppl/DC1 Listen to the Podcast interview] | |||
** [http://www.nytimes.com/2013/11/21/science/two-surprises-in-dna-of-boy-found-buried-in-siberia.html?hp NYT Report on Nov 20, 2013] | |||
* Workshop Assignment 1 (20 pts, which counts towards your '''active''' participation in the last two sessions). [Finalized 11/23 at 11am] Answer the following questions based on your class notes. Use complete sentences. | |||
# '''Genetic analysis of Brown Bear populations''' (pg 73-77). Your map should have shown that the four haplotype clades separate into four regional populations with only one exception (2 clades coexist at location "M"). One possible cause of such geographic separation of haplotypes is female philopatry (see pg.77). Explain how sequencing the Y chromosomes from the bears could support or dis-approve the female philopatry hypothesis. Structure your paragraph in the following way: | |||
## Start by explaining the philopatry hypothesis and how it may explain the observed distribution of mitochondrial haplotypes | |||
## If Y-chromosome haplotypes are more mixed in each region, is female philopatry hypothesis supported or disapproved? | |||
## If Y-chromosome haplotypes are just as geographically distinct as mitochondrial haplotypes, is female philopatry hypothesis supported or disapproved? | |||
## If the female philopatry is disapproved by Y-chromosome haplotypes, describe a possible explanation [Hint: Re-phrase one of the first two bullet points on pg.77] | |||
## Conclude with how geographic distribution of Y-chromosome haplotypes would support or dis-approve the female philopatry hypothesis | |||
<pre style="white-space: pre-wrap;">[The following answer is contributed by a student in the class] | |||
1. One possible explanation for the current geographic separation of haplotypes in brown bear population in northwest North America is the female philopatry hypothesis. This means female bears tend to live and breed within home ranges that are smaller than male bears and daughter bears often set up home ranges within their mother’s home range. | |||
2. If data on Y-chromosome haplotypes are more mixed in each region, this would support the female philopatry hypothesis. The reason for the difference in the pattern of geographical distribution is because male bears have larger home ranges than female bears and would disperse greater distances when breeding. A mixture of Y-chromosome haplotypes in each region would indicate greater gene flow between regional populations due to the tendency of male bears to roam. | |||
3. If data on Y-chromosome haplotypes are just as geographically distinct as mitochondrial haplotypes, this would disapprove the female philopatry hypothesis. This would indicate both maternal and paternal inheritance is restricted to each regional population. The philopatric behavior of female bears does not explain similar patterns of geographical distribution of Y-chromosome haplotypes. Therefore, there must be another reason behind the reproductive separation of each regional population of brown bears. | |||
4. In the event the female philopatry hypothesis is disapproved by data on Y-chromosome haplotypes, another possible explanation for the distinct haplotype classes in regional population is the founder effects hypothesis. This hypothesis believes that the brown bear population was fragmented due to climate changes in the past (associated with glaciations). This fragmentation also coincided with the shrinkage in population size due to the same reasons, leaving small pockets of the population that survived. The surviving members of these pockets may be referred to as “founders,” from whom the current population descended. The distinct haplotype classes found in each regional population may be the result of a genetic signature that is a legacy of each “founder” population of the region. | |||
5. In conclusion, the addition of sequencing Y-chromosome haplotypes would either help support the female philopatry hypothesis by showing mixed patterns of geographical distribution of haplotypes. Or disapprove the hypothesis by showing a similar pattern of geographical distribution of distinct haplotypes in each regional population. Either way, the additional experiment on the Y-chromosome would be a powerful tool in evaluating the female philopatry hypothesis. | |||
</pre> | |||
# '''Mal'ta Boy and origin of Native Americans''' (see links above). The Mal'ta Boy genome suggests that European sequence types in Native Americans are pre-historic and not post-Columbus. Describe evidence supporting this conclusion. Structure your paragraph in the following way: | |||
## Start with the main question ("source of European DNA in Native American genomes") and the previous theory [Hint: "post-Columbus gene flow"] | |||
## Describe the expected genetic composition of Native American genomes under the hypothesis of origin ''after'' the interbreeding between the West Eurasian and East Asian populations | |||
## Describe the expected genetic composition of Native American genomes under the hypothesis of origin ''before'' the interbreeding between the West Eurasian and East Asian populations | |||
## Explain why the Mal'ta Boy genome support the "after-interbreeding" hypothesis, using evidence of its mitochondrial and Y-chromosome haplotypes. | |||
## Conclude by describing how the Mal'ta Boy genome revised the theory on the geographic and genetic origins of Native Americans. | |||
<pre style="white-space: pre-wrap;">[The following answer is contributed by a student in the class] | |||
1. The source of European DNA in Native American genomes has long been assumed to occur after Christopher Columbus introduced the new world to Europe and is a result of the interbreeding between the native population and Europeans after the colonization of the Americas (circa 1492). However, the recent revelation of the genetic identity of the Mal’ta boy (which is closely related to Native Americans) seems to suggest the source of European DNA present in Native Americans are pre-historic and not post-Columbus, as we had assumed. In analyzing the genetic identity of the Mal’ta Boy and Native Americans, we may draw two hypotheses on the origin of Native Americans. The first hypothesis suggests that the ancestral population of Native Americans migrated to North American after the interbreeding between West Eurasian and East Asian populations. The second hypothesis suggests this ancestral population migrated before the interbreeding took place. | |||
2. In the scenario of the first hypothesis, we would expect the genetic composition of Native American genomes to be a mixture of West Eurasian and East Asian populations. Also, as descendents of this mixture, the Native American genome should be homogenous in the distribution of haplotypes that may be unique to this mixture. | |||
3. In the scenario of the second hypothesis, we would expect the genetic composition of Native American genomes to resemble either West Eurasian or East Asian. If the migration of both populations occurred prior to interbreeding, there should be pockets of Native American populations that reflect “founder effects,” having haplotypes unique only to West Euroasians or East Asians. | |||
4. By examining the Mal’ta Boy’s genome, it would seem the “after-interbreeding” hypothesis on the origin of Native Americans is much more plausible. First, both mitochondrial DNA and Y-chromosome indicate the Mal’ta Boy’s genes come from Europe and West Asia. We may conclude he is part of a regional population that has a steady gene flow of the mixed DNA of Europeans and West Asian. This is important as both his maternal and paternal inheritance reflects this mixture of West Eurasian. Second, his autosomal DNA shows he is closed related to Native Americans. Since the Mal’ta boy is dated at 24,000 years old and Native Americans are identified as approximately 15,000 year old, he is most likely an early ancestor of the Native American population. Lastly, the absence of East Asian DNA in the Mal’ta Boy strongly suggests that although he is an early ancestor, he is not part of the ancestral population that migrated to North America. The two-third proportion of East Asian DNA present in the Native America genome was most liked introduced to the Mal’ta Boy’s West Eurasian population and only after interbreeding were the ancestral population of Native America created. | |||
5. In conclusion, the revelation of Mal’ta Boy genome offers new insights to the genetic makeup and migration patterns of the first Americans that may revise our current theories on the origin of Native Americans. Mal’ta Boy provides genetic evidence that would suggest (1) the source of European DNA present in Native Americans was most like pre-historic and not post-Columbus and (2) there was an ancestral population of Native Americans that migrated to North America by crossing the Beringia carrying both European and East Asian DNA. | |||
</pre> | |||
===November 26. Genome Evolution (3)=== | |||
* Lecture Slides: [[File:Session-10.pptx|thumbnail]] | |||
* Workshop III. Tree of Life | |||
* Workshop Assignment 2 (15 pts). [Finalized 12/1, 12 noon] Answer the following questions '''with explanations'''. | |||
[[File:Tree-of-great-apes.jpg|thumbnail|right|Ape phylogeny]] | |||
# With reference to the tree at right, determine whether each of the following statement is True or False. Explain why. [Hint: Print out the tree and label each internal nodes] | |||
## Chimps are more related to Gorilla than to Human | |||
## Gorrilla is more related to the Common Chimp than to Human | |||
## A gene sequence from Gorrila is expected to be more similar to its human homolog (i.e., a homologous gene) than to its Orangutan homolog | |||
<pre style="white-space: pre-wrap;">[The following answer is contributed by a student in the class] | |||
1. Chimps are more related to Gorilla than to Human: False. Chimps are more related to human than to gorilla. Chimps and human’s most recent common ancestor is at node labeled B, about 6 mya. Chimps and gorilla’s most recent common ancestor is at node C, about 11 mya. Chimps and humans share a more recent common ancestor than chimps and gorillas, and relatedness is determined by the closeness of the shared ancestor. | |||
2. Gorilla is more related to the Common Chimp than to Human: False. Gorilla is equally related to common chimp and human. Node C is the location for the common ancestor of gorilla and common chimp, as well as for gorilla and human. | |||
3. A gene sequence from Gorrila is expected to be more similar to its human homolog (i.e., a homologous gene) than to its Orangutan homolog: True. Gorillas are more closely related to humans than to orangutans. The most recent shared ancestor of gorillas and orangutans is at node D (about 15 mya), while the most recent shared ancestor of gorillas and humans is at node C (about 11 mya). Two species that are more closely related will have more similarities in gene sequences, so the gorilla’s gene sequence will be more similar to its human homolog than to its orangutan homolog. | |||
</pre> | |||
# Describe the Tree of Life, including (a) three domains of life, (b) does it imply a single or multiple origins of life on Earth?, (c) at least one line of genetic evidence supporting the common origin of life. | |||
<pre style="white-space: pre-wrap;">[The following answer is contributed by a student in the class] | |||
The tree of life is used to determine species relatedness. There are three types of nodes on the tree: the Root is at the beginning of the tree (which is at the bottom of the above picture) and represents the most recent common ancestor of all species on the tree. Terminal nodes are at the ends and represent existing species. Internal nodes represent hypothetical ancestors after the root. Like the root, they are statistically inferred but may or may not have existed. Branches (lines) represent the number of changes between the ancestor and the descendent species; a longer branch indicates more changes in DNA. | |||
The three domains of life are: bacteria, archaea, and eukarya. Bacteria and archaea are unicellular and eukarya are multicellular. The tree of life implies a single origin of life on earth (the root of the tree is the common ancestor of all life forms). Evidence supporting the common origin of life includes the fact that the genetic code (the code of which triplets code for which amino acids) is universal. Similar core genes and similar DNA sequences among all species are also evidence for a common origin of life. | |||
</pre> | |||
# Based on the Tree of Life, explain why it is harder to design drugs to treat fungal diseases than to treat bacterial diseases. | |||
<pre style="white-space: pre-wrap;">[The following answer is contributed by a student in the class] | |||
Drugs designed for human use typically have selective toxicity—they target bacterial but not eukaryotic cells. Bacteria and Eukaryotes (the domain humans belong to) are two separate domains of life on the tree, so it is not difficult to find drugs that will affect one but not the other. There are ribosomal distinctions between bacteria and eukaryotes, so these drugs can target the ribosomes of bacteria and not humans to block protein synthesis. | |||
However, fungus and humans are both eukaryotes and have many genetic similarities, as seen on the tree of life. Fungus and animals are very closely related, sharing a quite recent common ancestor. There are therefore few distinctions between the ribosomes on fungus and humans, and it is very difficult to find a drug that will target only the fungus and not damage the human host. | |||
</pre> | |||
===December 3. Genome Evolution (4)=== | |||
* Lecture Slides: [[File:Session-11.pptx|thumbnail]] | |||
* Workshop IV. Human evolution | |||
* Workshop Assignment 3 (Finalized 12/7 1pm. 15 pts). | |||
[[File:Ch04f11.jpg|thumbnail]] | |||
# Based on sites #61-90 in the thioredoxin alignment (see figure at right), draft a phylogenetic tree of these four species. Show your work [Hint: Calculate pairwise differences. Then group two most similar species together. Proceed to adding the next-similar species. End by adding the least similar species.] | |||
# BLAST the [http://www.ncbi.nlm.nih.gov/nuccore/NC_005100.3?report=genbank&from=41824255&to=41824668 Rat cytC] gene sequence against the mouse genome. Based on the top hit, Identify the first five nucleotide sequence differences '''within the coding region'''. Are they synonymous or non-synonymous differences? Show your work. [Hint: identify the first 3-5 codons using the GenBank file. Then find the matching sequences in the BLAST output file.] | |||
# Build a molecular phylogeny of homologs of [http://www.ncbi.nlm.nih.gov/gene/672 human BRCA1 gene] using [http://www.phylogeny.fr www.phylogeny.fr website]. Show your work. [Hint: Follow the HomoloGene link to download protein sequences. Then use the phylogeny website to infer a tree. Use only species names for tip labels in your final tree.] | |||
<gallery align="center"> | |||
File:wkshop-assign3-1.png|Answer key for Q1 | |||
File:wkshop-assign3-2.png|Answer key for Q2 | |||
File:wkshop-assign3-3.png|Answer key for Q3 | |||
</gallery> | |||
===December 10. Review=== | |||
* Review Slides: [[File:Final-review.pptx|thumbnail]] | |||
===December 17. Final Cumulative Exam=== | |||
* 12:45-1:30: Paper-only exam | |||
* 1:30-2:10: Computer practicum | |||
==Links== | ==Links== | ||
* NCBI Human genome browser: [http://www.ncbi.nlm.nih.gov/projects/genome/guide/human/index.shtml NCBI Human Genome Page] | |||
* NIH Encode Project: [http://www.genome.gov/10005107#al-1 Discover functions in 1% human genome] | |||
* Smithsonian Exhibit: [http://unlockinglifescode.org/ ‘Genome: Unlocking Life’s Code’] | |||
* Should human genes be patented? [http://www.nytimes.com/2013/06/19/opinion/our-genes-their-secrets.html?hp An NYT Op-Ed] | * Should human genes be patented? [http://www.nytimes.com/2013/06/19/opinion/our-genes-their-secrets.html?hp An NYT Op-Ed] | ||
* Biological Databases on the Web: [http://www.oxfordjournals.org/nar/database/cap/ 2013 Collection by NAR] | * Biological Databases on the Web: [http://www.oxfordjournals.org/nar/database/cap/ 2013 Collection by NAR] | ||
* Biological Webservers: [http://nar.oxfordjournals.org/content/40/W1.toc 2012 Collection by NAR] | * Biological Webservers: [http://nar.oxfordjournals.org/content/40/W1.toc 2012 Collection by NAR] | ||
* The HapMap Project: [http://hapmap.ncbi.nlm.nih.gov/index.html.en Identifying major DNA variations in humans] | |||
* The 1000 Genomes Project: [http://www.1000genomes.org/ Identifying rare DNA variations in humans] | |||
* The UCSC Genome Browser: [http://genome.ucsc.edu/ Comparing human and other genomes] | |||
Latest revision as of 21:05, 10 December 2013
General Information

Course Description
- Background: A genome is the total genetic content of an organism. Driven by breakthroughs such as the decoding of the first human genome and rapid DNA-sequencing technologies, biomedical sciences are undergoing a rapid and profound transformation into a highly data-intensive field, which requires familiarity with concepts in both biology and computer science. Genome information is revolutionizing virtually all aspects of biology and medicine and will lead to major advances such as more efficient production of renewable energy, better cures for cancers, and longer and healthier life expectancy.
- Contents: This course will introduce genome-sequencing technologies, explore genome projects online, and discuss both the benefits and challenges (e.g., ethical and legal) of the genomic revolution to society. Each session will consist of lecture instructions and computer-based laboratory practices.
- Learning Goals
- Human Genome: Knowing the basic content and structure of the human genome
- Central Dogma: Understanding the Central Dogma and how genes are regulated (i.e., turned on/off)
- Genome diversity: Knowing the three major divisions of life, the use of DNA/protein sequences to classify organisms, and mechanisms of genome evolution
- Bioinformatics: Understanding the importance of computation and statistics in producing, analyzing, and disseminating genome information
- Learning Outcomes
- Be able to describe the structure and function of the human genome
- Be able to find chromosomal location, function, disease-association, and variability of a human gene using scientific databases
- Be able to identify and classify organisms based on their DNA sequences
- Be able to describe the processes of genome evolution, including mutation, duplication, recombination, horizontal gene transfer, genetic drift, and natural selection
- Textbook: Arthur Lesk (2012). Introduction to Genomics (2nd Edition). Oxford University Press. Amazon Link
- Academic Honesty: Hunter College regards acts of academic dishonesty (e.g., plagiarism, cheating on examinations, obtaining unfair advantage, and falsification of records and official documents) as serious offenses against the values of intellectual honesty. The College is committed to enforcing the CUNY Policy on Academic Integrity and will pursue cases of academic dishonesty according to the Hunter College Academic Integrity Procedures.
Grading Policy
- Treat assignments as take-home exams. Student performance will be evaluated by weekly assignments and three exams. While students are allowed to work in groups for assignments (and answers to some questions are available on the textbook website), students are expected to compose the final answers independently. Writings that are virtually exact copies to each other or to the web answers will be considered plagiarism. Zero points will be given to ALL involved individuals if the instructor considers there is enough evidence for plagiarism. To avoid being investigated for plagiarism, Do NOT copy or let others copy your work.
- Submit assignments in Printed Hard Copies. Email attachments will NOT be accepted. Each assignment will be graded based on timeliness (20%), effort (40%), and correctness (40%).
- Since each session will consist of a web-based practical session, attendance is required and part of the grade.
- Composition of your final grade
- Assignments: weekly assignments, totaling 100 pts
- Mid-term and Final Comprehensive Exams: 3 mid-terms X 50 points each = 150 pts
- Attendance and active classroom participation (50 pts): 1 unexcused absences = 40 pts; 2 absences = 30pts; More than 2 = 0. Points will be deducted for lack of participation in computer-lab exercises.
Weekly Schedule (All Tuesdays)
September 3. Introduction to Genomics (1)
- Lecture Slides:
- Course Overview
- Contents of the human genome (Chapter 1, pg. 3-10)
- Genome organization (Chapter 1, pg. 10-17)
- Assignment 1 (15 pts, due 9/10) [Finalized on 9/5, 7pm]
- (2 pts) (Note: modified from the textbook) Exercise 1.1. (a) Make a very rough estimate of the average density (e.g., number of genes per million base pairs) of protein-coding genes in the human genome, assuming total genome size ~ 3 x 10^6 bp and total number of genes ~ 3 X 10^4 genes.
Answer: 3x10^4 (genes)/3x10^9 (bp; typo in the original question)=1 gene every 10,000 bp
(b) Further assuming that the average gene length is 1000 bp, what is the proportion of bases that code for proteins?Answer: 3x10^4x10,000 (bp in genes)/3x10^9 (bp, total)=1x10^-2, or 1% of the total bases code for genes
- (4 pts) Exercise 1.3. For the standard genetic code (see lecture slides), give an example of a pair of codons related by a synonymous single-site substitution (a) at the third position and (b) at the first position. (c) Give an example of a pair of codons related by a non-synonymous single-site substitution at the third position. (d) Can a change in the second position of a codon ever produce a synonymous mutation?
Answer to (d): No. All 2nd position changes are non-synonymous. Stop codons are not "synonymous" amino acids.
- (4 pts) Exercise 1.4. (a) Is it possible to convert Phe to Tyr by a single base change? If so, what would be possible wild-type and mutant codons?
Answer: Yes. Either TTT (Wild Type, WT) to TAT (Mutant, MU), or TTC (WT) to TAC (MU). Both are changes at the 2nd codon position.
(b) Is it possible to convert Ser to Arg by a single base change? If so, what would be possible wild-type and mutant codons?Answer: Yes. Either AGT (Wild Type, WT) to CGT (Mutant, MU), or AGC (WT) to CGC (MU). Both are changes at the 1st codon position.
(c) What is the minimum number of base substitutions that would convert Cys to Glu?Answer: Three. Cys codons are either TGT or TGC, and Glu codons are either GAA or GAG. These two amino acids don't share the first, second, or the third codon positions.
(d) In the evolution off an essential protein encoded by a single gene, a Trp is converted to a Gln by two successive single-base changes. What is the intermediate codon?Answer: TGG (Trp) through CGG (Arg, 1st position change) to CAG (Gln, 2nd position change). The other possibility is highly unlikely since it goes through a stop codon: TGG through TAG (Stop, 2nd position change) to CAG (Gln, 1st position change)
- (5 pts) Weblem: Use the NCBI Human Genome Resource webpage to identify diseases associated with heamoglobin alpha-1 (HBA1). Summarize its association with the disease "alpha-thalassemia". Include key references and a discussion on why this is a case against "genetic determinism".
Answer: [http://www.omim.org/entry/141800 Online Mendelian Inheritance in Man database Entry 141800] lists 5 diseases associated with HBA1 mutations. See [http://www.omim.org/entry/604131 See OMIM Record #604131] for information on alpha-thalassemia. The key reference is Weatherall (2001), which concludes that heritability of alpha-thalassemia is low and environmental factors influences the disease outcome. Mutations at HBA1 locus alone is not a sufficient predictor of the disease, an argument against "genetic determinism" (At the least, it's not a single-gene genetic disease).
- (2 pts) (Note: modified from the textbook) Exercise 1.1. (a) Make a very rough estimate of the average density (e.g., number of genes per million base pairs) of protein-coding genes in the human genome, assuming total genome size ~ 3 x 10^6 bp and total number of genes ~ 3 X 10^4 genes.
September 10. Introduction to Genomics (2)
- Lecture Slides:
- Assignment 2 (Finalized on 9/11 @11pm)
- (2 pts) (Modified from Exercise 1.8). Using the NCBI human genome browser, find out which of the following bands of human chromosome 16 are gene-rich: p13.3, q22.1, or q11.2?
Answer: According to the NCBI Human Genome Resource page (at the 1/100 zoom level and using "recenter"), the q22.1 has 32 genes, q11.2 has 0 genes, and p13.3 has 74 genes. So q13.3 and q22.1 (both light-colored) are gene-rich, while q11.2 (darker-colored) is gene-poor.
- (4 pts) (Modified from Exercise 1.11). Make a printout of the GenBank file with the Accession "V00488" (print ONLY the part with the DNA sequence). Mark the following sequence regions with highlighters: (a) all introns, (b) all exons, (c) 5'-UTR and 3'-UTR, (d) start and stop codons. (Hint: Use the "Graphics" view with zooming.)
Answer: Using the Graphics view and zoom in at the nucleotide level, you will find the following coordinates: Primary transcript: 98-929, Exon 1: 98-230 (most likely 98-229), Exon 2: 348-551 (should be 347-551), Exon 3: 692-929, Intron 1: 231-347 (should be 230-346, starting with GT and ending in AG), Intron 2: 552-691, 5, Start Codon: 135-137, Stop Codon: 818-820, 5'-UTR: 98-134, 3'-UTR: 821-929
- (4 pts) (Modified from Exercise 1.12). On a separate printout of the GenBank file with the Accession "V00488" (print ONLY the part with the amino-acid sequence). Mark the following regions with highlighters: (a) amino acids coded for by exon 1, (b) by exon 2, (c) by exon 3. (Hint: Use the "Graphics" view with zooming). (D) Is it possible that a single codon is split by an intron (e.g., 1st codon position on exon 1 and 2nd & 3rd nucleotides on exon 2) ?
- (2 pts) (Modified from Exercise 1.8). Using the NCBI human genome browser, find out which of the following bands of human chromosome 16 are gene-rich: p13.3, q22.1, or q11.2?

Answer key for 1.11

Answer key for 1.12

September 17. Introduction to Genomics (3)
- Lecture Slides:
- Genome database (Chapter 1, pg.24-30)
- Cancer genomics: The Cancer Genome Atlas (TCGA) Project
- Genome evolution (Chapter 1. pg.30-35)
- Assignment 3 (Finalized on 9/18, Thursday, 11:55pm)
- (6 pts). Problem 1.6 (on page 38). Instructor's Note: (1) Mark your answers on a printout or copy of Figure 1.17(a); (2) for part (d), show your work. A simple choice without reasoning or explanation will NOT get full credits.
- (4 pts). Read/Watch the following links and extract one example to describe how genome sequencing helps health care.
September 24. First Mid-term Exam
- Review session: 12:45-1:40
- Exam Part I. 1:45-2:30. No computer access
- Exam Part II. 2:30-3:15. Computer-based Practicum (NCBI websites only)
October 1. Genome Structure and Function (1)
- Lecture Slides: (updated)
- Classic Genetics (Chapter 3, pg.80-82)
- Structure of DNA (Chapter 3, pg. 90-93)
- DNA replication and PCR (Chapter 3, pg 93-95)
- Assignment 4 (Finalized as of 10/2, 1pm)
- (5 pts). Two alleles (T and t) are segregating at a single genetic locus In a population of diploid individuals. "T" is the dominant allele and "t" is recessive for a phenotype (e.g., "Taster" and "Non-taster"). If two parents are both heterozygotes at this locus, what is the expected Mendelian ratio of genotypes of their offspring? Mendelian ratio of the phenotypes? Show your work.
Genotypes: TT:Tt:tt=1:2:1; Phenotypes: Taster:Non-taster=3:1
- (5 pts) Answer the following questions about the Polymerase Chain Reaction (PCR):
- Describe the three steps in each round of PCR, including the approximate temperature and the reaction occurring at each step
1. Denature DNA into single strands (95ºC); 2. Anneal primers (50ºC); 3. Synthesize DNA (72ºC)
- What are the key chemical ingredients required for amplifying a DNA region using PCR?
DNA template, primers, DNA polymerase, and nucleotides
- Starting from a single DNA molecule (note: double-stranded), what is the total number of copies of a gene on this DNA after 20 rounds of PCR amplification? Show your work.
2^20=1,048,576
- Describe the three steps in each round of PCR, including the approximate temperature and the reaction occurring at each step
- (5 pts) Fill in Table 1 on Slide #12 based on the gel picture. Explain the basis of your counts and frequencies.
- (5 pts). Two alleles (T and t) are segregating at a single genetic locus In a population of diploid individuals. "T" is the dominant allele and "t" is recessive for a phenotype (e.g., "Taster" and "Non-taster"). If two parents are both heterozygotes at this locus, what is the expected Mendelian ratio of genotypes of their offspring? Mendelian ratio of the phenotypes? Show your work.

October 8. Genome Structure and Function (2)
- Lecture Slides: (updated)
- Test of PTC genotype-phenotype association
- Linkage Map (Chapter 3, pg.82-85)
- Automated Sequencing (Chapter 3, pg 97-104)
- Assignment 5.(Finalized on Friday, 10/11/2013, 12 noon; Updated o nTuesday 10/15/2013)
- (2 pts) Exercise 3.2 (pg.110). Explain.
D=A1B1 x A2B2 - A1B2 x A2B1=0.2x0.45-0.15x0.2= 0.06. D is a measure of linkage disequilibrium (LD) between two loci. When D=0, it means that there is no association between allele frequencies on two loci, or no LD. In this case, D>0, suggesting that chromosome with A1B1 and A2B2 are over-represented (with respect to expectation) in the population.
- (3 pts) Exercise 3.6 (pg.110). Explain.
The bottom band in the middle lane corresponds to the 4 kb BamHI fragment. The 2nd band from the top in the right lane corresponds to the smallest (1.5 kb, with lowest molecular weight) EcoRI fragment. In a gel electrophoresis, DNA molecules travel from the negative electrode towards the positive electrode. This is because DNA is negatively charged in neutral conditions. Smaller DNA fragments, which have lower molecular weights, travel faster than longer ones. Note: 1 kb = 1 kilobases = 1,000 base pairs
- (5 pts) Problem 3.6 (pg.113). Explain. Hints: See figure at right for an illustration of the RNA stem and loop . Compare your results with those produced by this web-based RNA structure predictor: RNAfold web server
(a) DNA synthesis occurs in the direction of 5' to 3', so the shortest synthesized fragment is at the bottom. Therefore, the sequence is: 5'-CCTTATCTCTTGATAGGGAACCGGTTTCCCGGC-3'. (b) There is no long (6 bp or more) hairpin structure predicted for this sequence. The longest exact self-complementary region predicted by the RNAfold program is a 4-bp stem near the 3' end: 5'-CCTTATCTCTTGATAGGGAA{CCGG}TTTC{CCGG}C-3'. (c). Based on the previous analysis, this region of DNA is more likely to encode protein. - (5 pts) Perform a hypothetical genotype-phenotype association study. Individuals are categorized depending on having a disease ("Case") or not ("Control"). Each individual is genotyped at one SNP site with two possible states (e.g., "A" or "G"). The following table is a 2X3 contingency table from the study. Perform a chi-squared test using this website. Answer the following questions:
- What are the expected counts if there is no association between the genotype and the disease phenotype? (Null hypothesis)
See Table 2 below
- What is your chi-square value and the p value? What is your interpretation of the p value? Using a cutoff of p=0.05, what can you conclude from the study (i.e., does it show a statistically significant association between genotype and disease?)
chi-square = 8.97, probability (p value) = 0.011, which is the probability of obtaining observed counts (including more biased counts than these) by chance (i.e., if there is no association between the genotypes and the disease). If p<0.05 is considered statistically significant, the result suggest that the null hypothesis ("no association") could be rejected and there is a statistically significant association between the genotype and the disease. - According to your comparisons between observed and expected counts, which genotype is the most over-represented in individuals with diseases? Which genotype is the most under-represented with diseases?
Compare the observed counts (in Table 1) with corresponding expected counts (Table 2). The "GG" genotype is the most over-represented and the "AA" genotype is the most under-represented in the disease cases.
- What are the expected counts if there is no association between the genotype and the disease phenotype? (Null hypothesis)
- (2 pts) Exercise 3.2 (pg.110). Explain.

| AA | AG | GG | |
|---|---|---|---|
| Case | 221 | 198 | 55 |
| Control | 279 | 212 | 35 |
| AA | AG | GG | |
|---|---|---|---|
| Case | 237 | 194 | 43 |
| Control | 263 | 216 | 47 |
October 15. No Class (Monday Schedule)
October 22. Genome Structure and Function (3)
- Lecture Slides:
- Virtual Lab: Microarray
- Videos for understanding genome functions:
| Transcription | Splicing | Translation |
|---|---|---|
| WsofH466lqk|300|center|Transcription}} | FVuAwBGw_pQ|300|center|Splicing}} | 5bLEDd-PSTQ|300|center|Translation}} |
- Assignment 6 (10 pts). Use the Cancer Transcriptome Website for the following exercises:
- Access the expression data for protein kinase C Beta (PRKCB1).
- Name a group of cancers associated with underexpression of PRKCB1. Attach the figure and show your rationale.
Green color indicates under-expression of a gene in cancer cells (relative to control, healthy cells). Figure 1 (see below) shows that PRKCB1 is under-expressed in, e.g., colon and breast cancer cells.
Notice: Flipped answers to this and the next question are acceptable since the color scheme is not clearly specified on the website. - Name a group of cancers associated with overexpression of PRKCB1. Attach the figure and show your rationale.
Red color indicates over-expression of a gene in cancer cells (relative to control, healthy cells). Figure 1 (see below) shows that PRKCB1 is over-expressed in leukemia cells.
- Name 3 genes with similar cancer expression patterns as PRKCB1. Attach the figure and show your rationale.
Figure 2 (see below) shows that PLCE, CD53, PTPRC, CDW52, LRMP, RHAG, and PDCD genes have similar expression patterns among cancer cells as PRKCB1.


October 29. Genome Structure and Function (4)
- Lecture Slides:
- Cancer Genomics: Field trip to 3rd Annual Symposium of Systems Biology of Cancer at MSKCC. We will attend the Afternoon Session B, which has three talks during 1:30-3pm. Take notes and hand in one copy
- Assignment #7. (Finalized Wed, 10/30, 3pm)
- (2 pts) Exercise 3.14 (pg. 111). Show your work.
see "Contig 2" in the following figure
- (3 pts) Exercise 3.15 (pg. 111). Show your work. [Hint: Enlarge the picture (e.g., double the size), and then measure the lengths of individual fragments and the region with a ruler. Read the measurements in millimeters.]
(a) The minimum coverage of any position is zero (in the gap region, see figure at right), (b) the maximal coverage of any position is at the red vertical line (see figure at right), which is covered by 9 individual reads. (c) The average coverage of the entire region is the sum of lengths of all reads divided by the length of the region. The answer is approximately 4, indicating that on average each DNA position is covered by 4 sequencing reads.
- (5 pts) Search the NCBI Gene database for "BRCA1" gene. Based on this Gene Card for the human version of the gene, summarize its molecular function, chromosomal location, gene structure (number of exons and introns, lengths of primary transcript, mRNA, and protein), mutations associated with cancers. Follow the link to OMIM to find out what are the 185delAG and 5382insC mutations (base substitution, deletion, insertion, consequence to protein synthesis?) and in what exons do they appear.
- Molecular function: "This gene encodes a nuclear phosphoprotein that plays a role in maintaining genomic stability, and it also acts as a tumor suppressor...This protein thus plays a role in transcription, DNA repair of double-stranded breaks, and recombination."
- Chromosomal location: Chromosome 17: 17q21
- Gene structure: Five splicing variants (or isoforms) of BRCA1 are recorded in this GenBank file. For example, Isoform 4 has a primary transcript from base #13,546 to 93,366 (a total of 93366-13546+1=79,821 bases), 21 introns, 22 exons, a protein sequence of 759 amino acids (see protein_id="NP_009229.2"), an mRNA with 3 times protein length, plus 5'- and 3'-UTRs (approximately 3 x 759=2,277 bases).
- Mutations associated with cancers: "Mutations in this gene are responsible for approximately 40% of inherited breast cancers and more than 80% of inherited breast and ovarian cancers. " See two examples below.
- These are deletion/insertion (indel) mutations in exons and thus cause frame shifts and lead to truncated proteins. The 185delAG mtuation is a "2-bp (AG185) deletion in the normal sequence TTA GAG of codons 22-23 in exon 3. The AGAG presumably predisposed to the deletion. This mutation changes the reading frame of the mRNA and causes a premature termination codon at position 39." The 5382insC mutation is "a 1-bp (C) insertion at position 5382 in exon 20, changing the reading frame of the mRNA and causing a premature termination codon at position 1829 in exon 24."

[Hint: See following youtube video for different types of mutations: {{#ev:youtube|kp0esidDr-c|300|center|Frameshift mutations}}
November 5. 2nd Mid-term Exam
- 12:45-1:45. Review
- 1:45 - 2:30. Exam without computer access
- 2:30 - 3:15. Computer-based practicum
November 12. Genome Evolution (1)
- Lecture Slides:
- Workshop I. Molecular population genetics and classification (Chapter 2)
- (5 pts) Assignment 8 (Finalized on 11/14, 3pm). Using haplotype sequences in Table 2.2 (pg 75), compute sequence differences between each pair of Clade I, II, III, and IV. Within each clade, use the last sequence as the representative. Your answers should include the following items (and show your procedures, not just the answers):
- Copy and paste of four chosen sequences
- Counts of nucleotide differences between each pair of sequences (a total of 6 pairs)
- Fraction of sequence differences for each pair, using the total length of 294 bases
- Estimates of divergence time (in years) between each pair of sequences, assuming a sequence-divergence rate of 0.125 changes per 10,000 years.
Hint: Present your answers using tables like this one:
| Clade I | Clade II | Clade III | Clade IV | |
|---|---|---|---|---|
| Clade I | 0 | 11 base differences; 11/294=0.0374 diff per base; 0.0374/0.125 x 10,000=2993 years of divergence | 11;0.0374; 2993 | 20; 0.0680; 5442 |
| Clade II | (blank) | 0 | 18; 0.0612; 4898 | 23; 0.0782; 6259 |
| Clade III | (blank) | (blank) | 0 | 23; 0.0782; 6259 |
| Clade IV | (blank) | (blank) | (blank) | 0 |
November 19. Genome Evolution (2)
- Lecture Slides:
- Workshop I. Molecular population genetics and classification (Chapter 2)
- Workshop II. Origin of Native Americans
- Workshop Assignment 1 (20 pts, which counts towards your active participation in the last two sessions). [Finalized 11/23 at 11am] Answer the following questions based on your class notes. Use complete sentences.
- Genetic analysis of Brown Bear populations (pg 73-77). Your map should have shown that the four haplotype clades separate into four regional populations with only one exception (2 clades coexist at location "M"). One possible cause of such geographic separation of haplotypes is female philopatry (see pg.77). Explain how sequencing the Y chromosomes from the bears could support or dis-approve the female philopatry hypothesis. Structure your paragraph in the following way:
- Start by explaining the philopatry hypothesis and how it may explain the observed distribution of mitochondrial haplotypes
- If Y-chromosome haplotypes are more mixed in each region, is female philopatry hypothesis supported or disapproved?
- If Y-chromosome haplotypes are just as geographically distinct as mitochondrial haplotypes, is female philopatry hypothesis supported or disapproved?
- If the female philopatry is disapproved by Y-chromosome haplotypes, describe a possible explanation [Hint: Re-phrase one of the first two bullet points on pg.77]
- Conclude with how geographic distribution of Y-chromosome haplotypes would support or dis-approve the female philopatry hypothesis
[The following answer is contributed by a student in the class] 1. One possible explanation for the current geographic separation of haplotypes in brown bear population in northwest North America is the female philopatry hypothesis. This means female bears tend to live and breed within home ranges that are smaller than male bears and daughter bears often set up home ranges within their mother’s home range. 2. If data on Y-chromosome haplotypes are more mixed in each region, this would support the female philopatry hypothesis. The reason for the difference in the pattern of geographical distribution is because male bears have larger home ranges than female bears and would disperse greater distances when breeding. A mixture of Y-chromosome haplotypes in each region would indicate greater gene flow between regional populations due to the tendency of male bears to roam. 3. If data on Y-chromosome haplotypes are just as geographically distinct as mitochondrial haplotypes, this would disapprove the female philopatry hypothesis. This would indicate both maternal and paternal inheritance is restricted to each regional population. The philopatric behavior of female bears does not explain similar patterns of geographical distribution of Y-chromosome haplotypes. Therefore, there must be another reason behind the reproductive separation of each regional population of brown bears. 4. In the event the female philopatry hypothesis is disapproved by data on Y-chromosome haplotypes, another possible explanation for the distinct haplotype classes in regional population is the founder effects hypothesis. This hypothesis believes that the brown bear population was fragmented due to climate changes in the past (associated with glaciations). This fragmentation also coincided with the shrinkage in population size due to the same reasons, leaving small pockets of the population that survived. The surviving members of these pockets may be referred to as “founders,” from whom the current population descended. The distinct haplotype classes found in each regional population may be the result of a genetic signature that is a legacy of each “founder” population of the region. 5. In conclusion, the addition of sequencing Y-chromosome haplotypes would either help support the female philopatry hypothesis by showing mixed patterns of geographical distribution of haplotypes. Or disapprove the hypothesis by showing a similar pattern of geographical distribution of distinct haplotypes in each regional population. Either way, the additional experiment on the Y-chromosome would be a powerful tool in evaluating the female philopatry hypothesis.
- Mal'ta Boy and origin of Native Americans (see links above). The Mal'ta Boy genome suggests that European sequence types in Native Americans are pre-historic and not post-Columbus. Describe evidence supporting this conclusion. Structure your paragraph in the following way:
- Start with the main question ("source of European DNA in Native American genomes") and the previous theory [Hint: "post-Columbus gene flow"]
- Describe the expected genetic composition of Native American genomes under the hypothesis of origin after the interbreeding between the West Eurasian and East Asian populations
- Describe the expected genetic composition of Native American genomes under the hypothesis of origin before the interbreeding between the West Eurasian and East Asian populations
- Explain why the Mal'ta Boy genome support the "after-interbreeding" hypothesis, using evidence of its mitochondrial and Y-chromosome haplotypes.
- Conclude by describing how the Mal'ta Boy genome revised the theory on the geographic and genetic origins of Native Americans.
[The following answer is contributed by a student in the class] 1. The source of European DNA in Native American genomes has long been assumed to occur after Christopher Columbus introduced the new world to Europe and is a result of the interbreeding between the native population and Europeans after the colonization of the Americas (circa 1492). However, the recent revelation of the genetic identity of the Mal’ta boy (which is closely related to Native Americans) seems to suggest the source of European DNA present in Native Americans are pre-historic and not post-Columbus, as we had assumed. In analyzing the genetic identity of the Mal’ta Boy and Native Americans, we may draw two hypotheses on the origin of Native Americans. The first hypothesis suggests that the ancestral population of Native Americans migrated to North American after the interbreeding between West Eurasian and East Asian populations. The second hypothesis suggests this ancestral population migrated before the interbreeding took place. 2. In the scenario of the first hypothesis, we would expect the genetic composition of Native American genomes to be a mixture of West Eurasian and East Asian populations. Also, as descendents of this mixture, the Native American genome should be homogenous in the distribution of haplotypes that may be unique to this mixture. 3. In the scenario of the second hypothesis, we would expect the genetic composition of Native American genomes to resemble either West Eurasian or East Asian. If the migration of both populations occurred prior to interbreeding, there should be pockets of Native American populations that reflect “founder effects,” having haplotypes unique only to West Euroasians or East Asians. 4. By examining the Mal’ta Boy’s genome, it would seem the “after-interbreeding” hypothesis on the origin of Native Americans is much more plausible. First, both mitochondrial DNA and Y-chromosome indicate the Mal’ta Boy’s genes come from Europe and West Asia. We may conclude he is part of a regional population that has a steady gene flow of the mixed DNA of Europeans and West Asian. This is important as both his maternal and paternal inheritance reflects this mixture of West Eurasian. Second, his autosomal DNA shows he is closed related to Native Americans. Since the Mal’ta boy is dated at 24,000 years old and Native Americans are identified as approximately 15,000 year old, he is most likely an early ancestor of the Native American population. Lastly, the absence of East Asian DNA in the Mal’ta Boy strongly suggests that although he is an early ancestor, he is not part of the ancestral population that migrated to North America. The two-third proportion of East Asian DNA present in the Native America genome was most liked introduced to the Mal’ta Boy’s West Eurasian population and only after interbreeding were the ancestral population of Native America created. 5. In conclusion, the revelation of Mal’ta Boy genome offers new insights to the genetic makeup and migration patterns of the first Americans that may revise our current theories on the origin of Native Americans. Mal’ta Boy provides genetic evidence that would suggest (1) the source of European DNA present in Native Americans was most like pre-historic and not post-Columbus and (2) there was an ancestral population of Native Americans that migrated to North America by crossing the Beringia carrying both European and East Asian DNA.
November 26. Genome Evolution (3)
- Lecture Slides:
- Workshop III. Tree of Life
- Workshop Assignment 2 (15 pts). [Finalized 12/1, 12 noon] Answer the following questions with explanations.

- With reference to the tree at right, determine whether each of the following statement is True or False. Explain why. [Hint: Print out the tree and label each internal nodes]
- Chimps are more related to Gorilla than to Human
- Gorrilla is more related to the Common Chimp than to Human
- A gene sequence from Gorrila is expected to be more similar to its human homolog (i.e., a homologous gene) than to its Orangutan homolog
[The following answer is contributed by a student in the class] 1. Chimps are more related to Gorilla than to Human: False. Chimps are more related to human than to gorilla. Chimps and human’s most recent common ancestor is at node labeled B, about 6 mya. Chimps and gorilla’s most recent common ancestor is at node C, about 11 mya. Chimps and humans share a more recent common ancestor than chimps and gorillas, and relatedness is determined by the closeness of the shared ancestor. 2. Gorilla is more related to the Common Chimp than to Human: False. Gorilla is equally related to common chimp and human. Node C is the location for the common ancestor of gorilla and common chimp, as well as for gorilla and human. 3. A gene sequence from Gorrila is expected to be more similar to its human homolog (i.e., a homologous gene) than to its Orangutan homolog: True. Gorillas are more closely related to humans than to orangutans. The most recent shared ancestor of gorillas and orangutans is at node D (about 15 mya), while the most recent shared ancestor of gorillas and humans is at node C (about 11 mya). Two species that are more closely related will have more similarities in gene sequences, so the gorilla’s gene sequence will be more similar to its human homolog than to its orangutan homolog.
- Describe the Tree of Life, including (a) three domains of life, (b) does it imply a single or multiple origins of life on Earth?, (c) at least one line of genetic evidence supporting the common origin of life.
[The following answer is contributed by a student in the class] The tree of life is used to determine species relatedness. There are three types of nodes on the tree: the Root is at the beginning of the tree (which is at the bottom of the above picture) and represents the most recent common ancestor of all species on the tree. Terminal nodes are at the ends and represent existing species. Internal nodes represent hypothetical ancestors after the root. Like the root, they are statistically inferred but may or may not have existed. Branches (lines) represent the number of changes between the ancestor and the descendent species; a longer branch indicates more changes in DNA. The three domains of life are: bacteria, archaea, and eukarya. Bacteria and archaea are unicellular and eukarya are multicellular. The tree of life implies a single origin of life on earth (the root of the tree is the common ancestor of all life forms). Evidence supporting the common origin of life includes the fact that the genetic code (the code of which triplets code for which amino acids) is universal. Similar core genes and similar DNA sequences among all species are also evidence for a common origin of life.
- Based on the Tree of Life, explain why it is harder to design drugs to treat fungal diseases than to treat bacterial diseases.
[The following answer is contributed by a student in the class] Drugs designed for human use typically have selective toxicity—they target bacterial but not eukaryotic cells. Bacteria and Eukaryotes (the domain humans belong to) are two separate domains of life on the tree, so it is not difficult to find drugs that will affect one but not the other. There are ribosomal distinctions between bacteria and eukaryotes, so these drugs can target the ribosomes of bacteria and not humans to block protein synthesis. However, fungus and humans are both eukaryotes and have many genetic similarities, as seen on the tree of life. Fungus and animals are very closely related, sharing a quite recent common ancestor. There are therefore few distinctions between the ribosomes on fungus and humans, and it is very difficult to find a drug that will target only the fungus and not damage the human host.
December 3. Genome Evolution (4)
- Lecture Slides:
- Workshop IV. Human evolution
- Workshop Assignment 3 (Finalized 12/7 1pm. 15 pts).

- Based on sites #61-90 in the thioredoxin alignment (see figure at right), draft a phylogenetic tree of these four species. Show your work [Hint: Calculate pairwise differences. Then group two most similar species together. Proceed to adding the next-similar species. End by adding the least similar species.]
- BLAST the Rat cytC gene sequence against the mouse genome. Based on the top hit, Identify the first five nucleotide sequence differences within the coding region. Are they synonymous or non-synonymous differences? Show your work. [Hint: identify the first 3-5 codons using the GenBank file. Then find the matching sequences in the BLAST output file.]
- Build a molecular phylogeny of homologs of human BRCA1 gene using www.phylogeny.fr website. Show your work. [Hint: Follow the HomoloGene link to download protein sequences. Then use the phylogeny website to infer a tree. Use only species names for tip labels in your final tree.]

Answer key for Q1

Answer key for Q2

Answer key for Q3



December 10. Review
- Review Slides:
December 17. Final Cumulative Exam
- 12:45-1:30: Paper-only exam
- 1:30-2:10: Computer practicum
Links
- NCBI Human genome browser: NCBI Human Genome Page
- NIH Encode Project: Discover functions in 1% human genome
- Smithsonian Exhibit: ‘Genome: Unlocking Life’s Code’
- Should human genes be patented? An NYT Op-Ed
- Biological Databases on the Web: 2013 Collection by NAR
- Biological Webservers: 2012 Collection by NAR
- The HapMap Project: Identifying major DNA variations in humans
- The 1000 Genomes Project: Identifying rare DNA variations in humans
- The UCSC Genome Browser: Comparing human and other genomes