Pseudomonas population genomics: Difference between revisions
Jump to navigation
Jump to search
imported>Rayrah |
imported>Rayrah |
||
| Line 17: | Line 17: | ||
## Protein ortholog data: [http://pseudomonas.com/alignPolymorphicGeneSequencesStep1.do?feature_id_parent=105676&start=1583956&stop=1584798&replicon_id_reference=136&alphabet=protein&limit_to_species=false fleN], [http://pseudomonas.com/alignPolymorphicGeneSequencesStep1.do?feature_id_parent=104960&start=1187587&stop=1189059&replicon_id_reference=136&alphabet=protein&limit_to_species=false fleQ], [http://pseudomonas.com/alignPolymorphicGeneSequencesStep1.do?feature_id_parent=105674&start=1582528&stop=1583817&replicon_id_reference=136&alphabet=protein&limit_to_species=false flhF] | ## Protein ortholog data: [http://pseudomonas.com/alignPolymorphicGeneSequencesStep1.do?feature_id_parent=105676&start=1583956&stop=1584798&replicon_id_reference=136&alphabet=protein&limit_to_species=false fleN], [http://pseudomonas.com/alignPolymorphicGeneSequencesStep1.do?feature_id_parent=104960&start=1187587&stop=1189059&replicon_id_reference=136&alphabet=protein&limit_to_species=false fleQ], [http://pseudomonas.com/alignPolymorphicGeneSequencesStep1.do?feature_id_parent=105674&start=1582528&stop=1583817&replicon_id_reference=136&alphabet=protein&limit_to_species=false flhF] | ||
## Protocol: | ## Protocol: | ||
### Fasta headers are too long for tree, run: [https://www.dropbox.com/s/x2p4joeqg7omfub/rename.pl rename.pl] to shorten names <code> | ### Fasta headers are too long for tree, run: [https://www.dropbox.com/s/x2p4joeqg7omfub/rename.pl rename.pl] to shorten names. Usage:<code> >rename.pl <FASTA_file> > <OUTPUTfilename.fas> </code> (will rewrite script to create automatic output file) | ||
### Next run [http://www.drive5.com/muscle/Alignment muscle] Usage: <code> > muscle -in <FASTA_File> -out <OUTPUTfilename.aln> -clwstrict(/code) Phylogeny; Tree display (R commands) | |||
{| class="wikitable" | {| class="wikitable" | ||
! Gene !! Alignment !! Tree !! Notes | ! Gene !! Alignment !! Tree !! Notes | ||
Revision as of 20:58, 18 June 2013
Projects
- Build a local genome database
- Database schema:
- "genome": genome_id, strain_name, ncbi_taxid
- "orf": genome_id, locus_tag, start, stop, strand, genome_name, product_name
- "orth_orf": orth_orf_id, locus_name, genome_id, orth_class
- Parsing scripts
- Rayees Parsing code, requires that you remove columns 9-27 using bash command:
cut -c 1-8(I will write a bash script that does this and runs the program) https://www.dropbox.com/s/lpxxbkxeyw7frrn/parser.pl
- Rayees Parsing code, requires that you remove columns 9-27 using bash command:
- Database loading scripts
- Database schema:
- Molecular Evolution of flagellum genes
- Download orthologs
- Reconstruct phylogenetic tree
- Run PAML tests
Update: June 18, 2013
- Material and Methods
- Protein ortholog data: fleN, fleQ, flhF
- Protocol:
- Fasta headers are too long for tree, run: rename.pl to shorten names. Usage:
>rename.pl <FASTA_file> > <OUTPUTfilename.fas>(will rewrite script to create automatic output file) - Next run muscle Usage:
> muscle -in <FASTA_File> -out <OUTPUTfilename.aln> -clwstrict(/code) Phylogeny; Tree display (R commands)
- Fasta headers are too long for tree, run: rename.pl to shorten names. Usage:
Gene
Alignment
Tree
Notes
fleN
fleN pep alignment
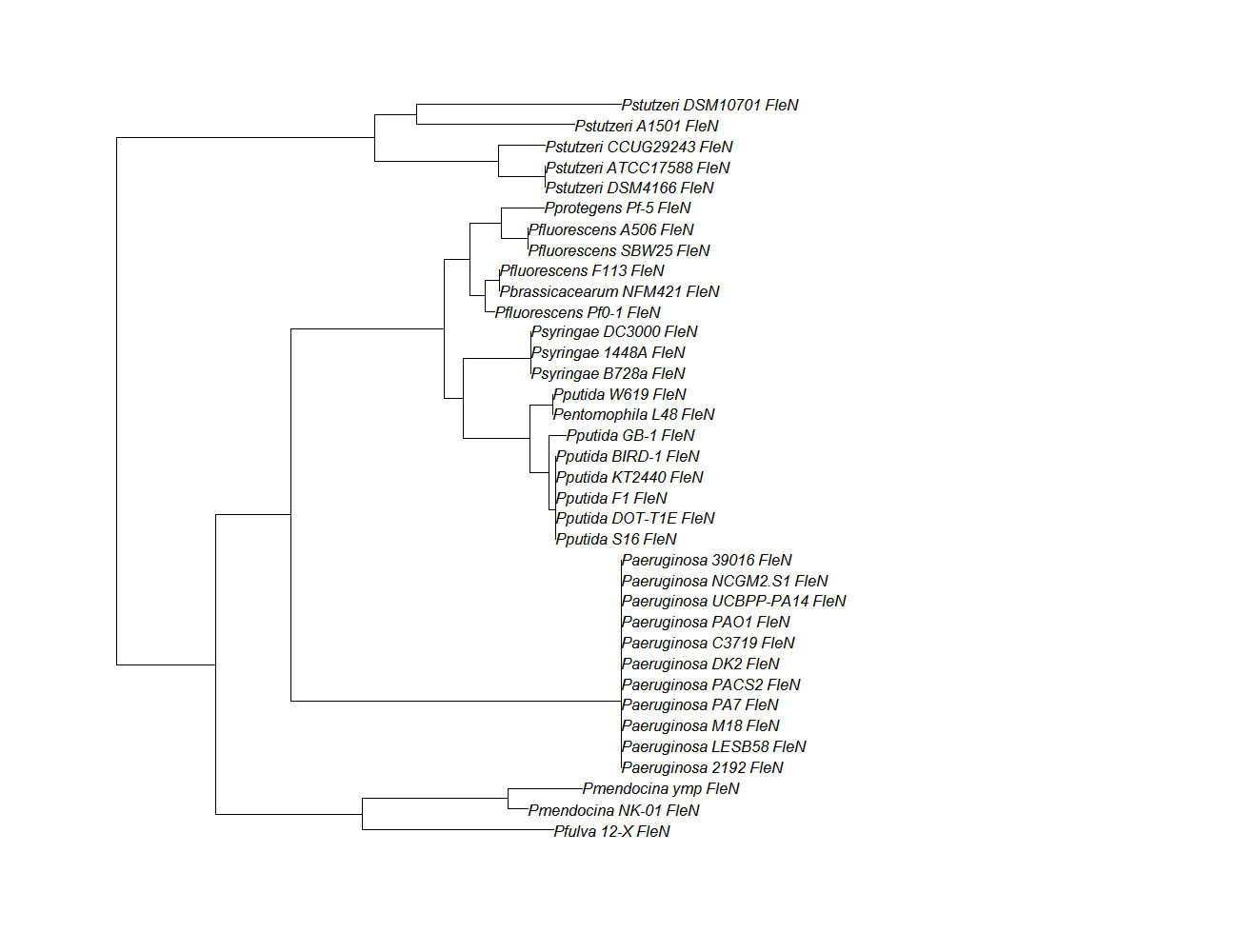
Conserved Domain
fleQ
fleQ pep alignment
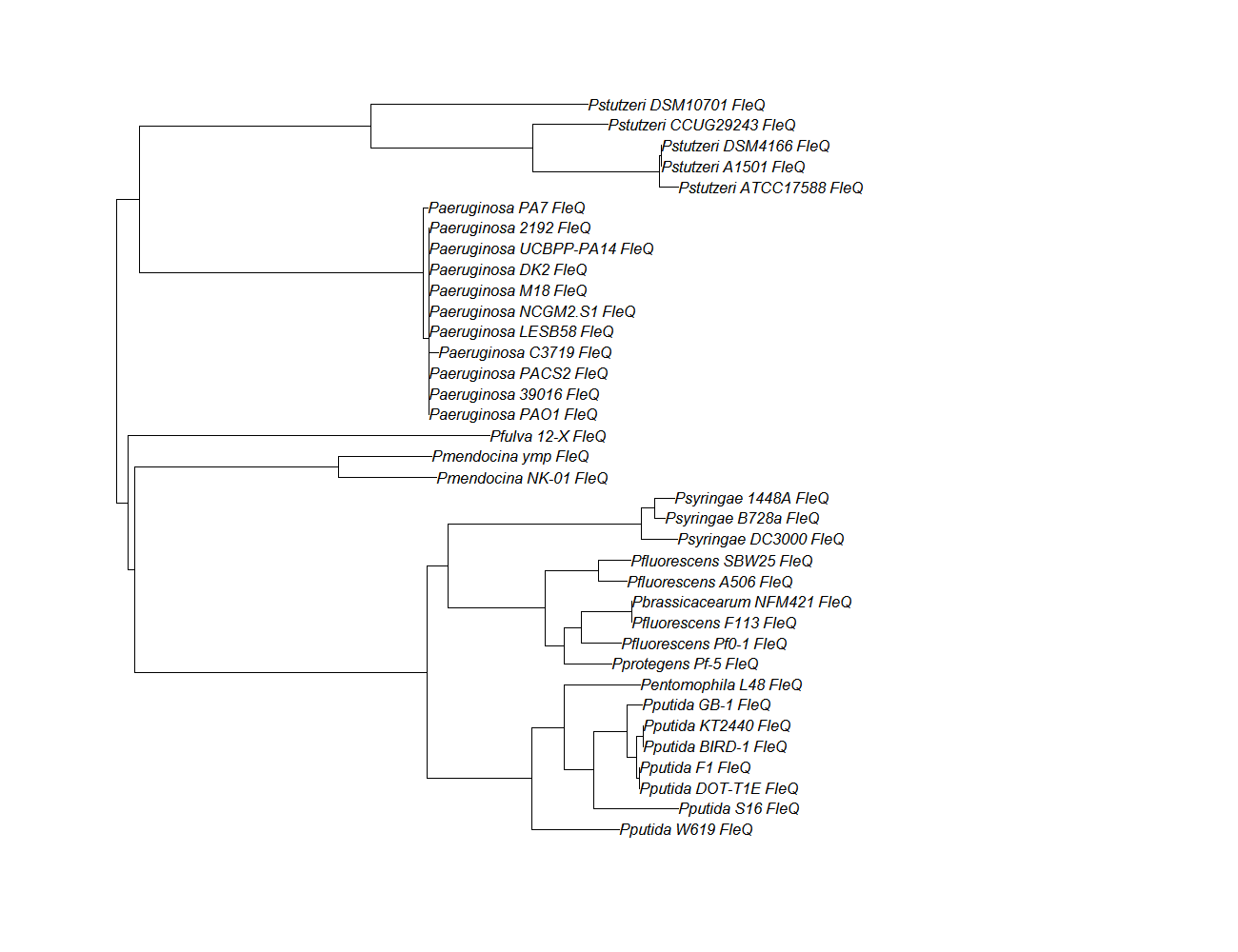
Conserved Domain
flhf
flhF pep alignment
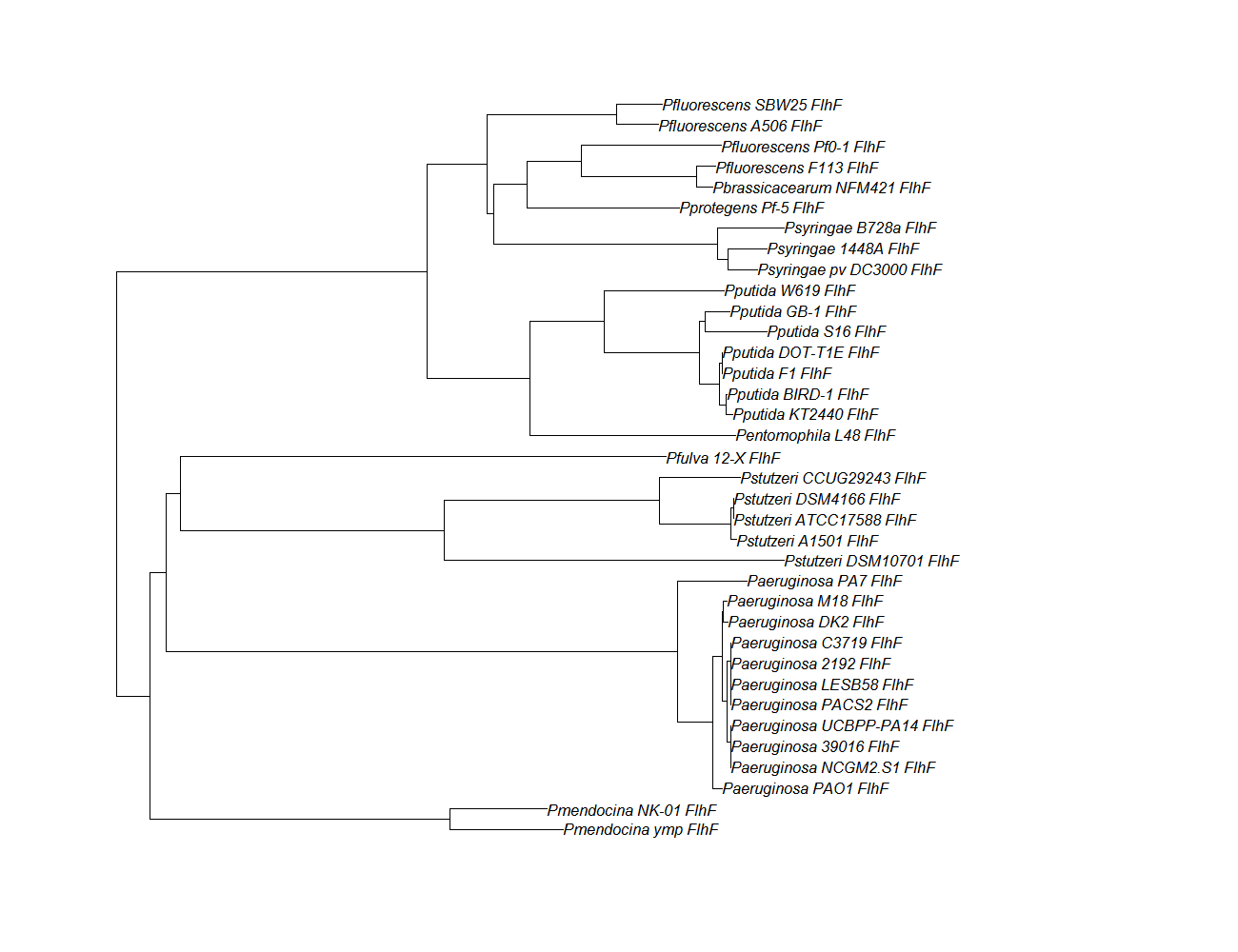
Conserved Domain



Benchmark: June 11, 2013
- Finish parsing the genome files to upload the "orf" table (Raymond & Rayees)
- Rayees Parsed genome files: https://www.dropbox.com/sh/k0zktvvmv39op9i/1zBercEky8
- Parsing the ortholog file to upload the "orth_orf" table (Raymond)
- Identify and download fleN, fleQ, and flhF orthologs & align them (Rayees)