Biol425 2013
Jump to navigation
Jump to search
General Information
Course Description
- Background: Biomedical research is becoming a high-throughput science. As a result, information technology plays an increasingly important role in biomedical discovery. Bioinformatics is a new interdisciplinary field formed between molecular biology and computer science.
- Contents: This course will introduce both bioinformatics theories and practices. Topics include: database searching, sequence alignment, molecular phylogenetics, structure prediction, and microarray analysis. The course is held in a UNIX-based instructional lab specifically configured for bioinformatics applications.
- Problem-based Learning (PBL): For each session, students will work in groups to solve a set of bioinformatics problems. Instructor will serve as the facilitator rather than a lecturer. Evaluation of student performance include both active participation in the classroom work as well as quality of assignments (see #Grading Policy).
- Learning Goals: After competing the course, students should be able to perform most common bioinformatics analysis in a biomedical research setting. Specifically, students will be able to
- Approach biological questions evolutionarily ("Tree-thinking")
- Evaluate and interpret computational results statistically ("Statistical-thinking")
- Formulate informatics questions quantitatively and precisely ("Abstraction")
- Design efficient procedures to solve problems ("Algorithm-thinking")
- Manipulate high-volume textual data using UNIX tools, Perl/BioPerl, R, and Relational Database ("Data Visualization")
- Pre-requisites: This 3-credit course is designed for upper-level undergraduates and graduate students. Prior experiences in the UNIX Operating System and at least one programming language are required. Hunter pre-requisites are CSCI132 (Practical Unix and Perl Programming) and BIOL300 (Biochemistry) or BIOL302 (Molecular Genetics), or permission by the instructor. Warning: This is a programming-based bioinformatics course. Working knowledge of UNIX and Perl is required for successful completion of the course.
- Textbook: Krane & Raymer (2003). Fundamental Concepts of Bioinformatics. Pearson Education, Inc. (ISBN 0-8053-4633-3)
- Academic Honesty: Hunter College regards acts of academic dishonesty (e.g., plagiarism, cheating on examinations, obtaining unfair advantage, and falsification of records and official documents) as serious offenses against the values of intellectual honesty. The College is committed to enforcing the CUNY Policy on Academic Integrity and will pursue cases of academic dishonesty according to the Hunter College Academic Integrity Procedures.
Grading Policy
- Treat assignments as take-home exams. Student performance will be evaluated by weekly assignments and projects. While these are take-home projects and students are allowed to work in groups and answers to some of the questions are provided in the back of the textbook, students are expected to compose the final short answers, computer commands, and code independently. There are virtually an unlimited number of ways to solve a computational problem, as are ways and personal styles to implement an algorithm. Writings and blocks of codes that are virtually exact copies between individual students will be investigated as possible cases of plagiarism (e.g., copies from the Internet, text book, or each other). In such a case, the instructor will hold closed-door exams for involved individuals. Zero credits will be given to ALL involved individuals if the instructor considers there is enough evidence for plagiarism. To avoid being investigated for plagiarism, Do NOT copy from others or let others copy your work.
- Submit assignments in Printed Hard Copies. Email attachments will NOT be accepted. Each assignment will be graded based on timeliness (10%), whether executable or having major errors (50%), algorithm efficiency (10%), and readability in programming styles (30%, see #Assignment Expectations).
- The grading scheme
- Assignments (100 pts): 10 exercises.
- Mid-term (50 pts).
- Final Project (50 pts)
- Classroom performance (50 pts): Active engagement in classroom exercises and discussions
- Attendance (50 pts): 1 unexcused absences = 40; 2 absences = 30; More than 2 = 0.
Assignment Expectations
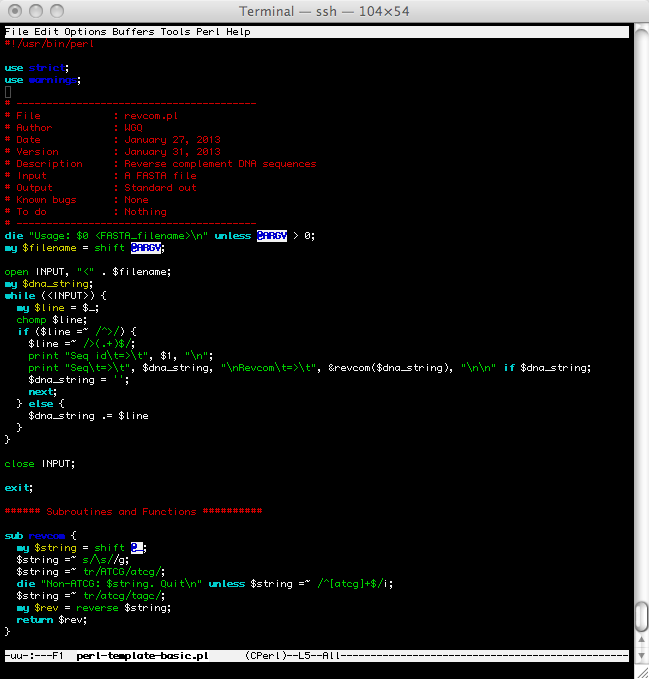
- Use a programming editor (e.g., vi or emacs) so you could have features like automatic syntax highlighting, indentation, and matching of quotes and parenthesis.
- All PERL code must begin with "use strict; and use warnings;" statements. For each assignment, unless otherwise stated, I would like the full text of the source code. Since you cannot print using the text editor in the lab (even if you are connected from home), you must copy and paste the code into a word processor or a local text editor. If you are using a word processor, change the font to a fixed-width/monospace font. On Windows, this is usually Courier.
- Also, unless otherwise stated, both the input and the output of the program must be submitted as well. This should also be in fixed-width font, and you should label it in such a way so that I know it is the program's input/output. This is so that I know that you've run the program, what data you have used, and what the program produced. If you are working from the lab, one option is to email the code to yourself, change the font, and then print it somewhere else as there is no printer in the lab.
- Recommended Style
- Bad Style
{kind=link}
{kind=link}
Course Schedule (All Wednesdays)
January 30. Browse Genome & Transcriptome Files with Unix Tools
- Course Overview
- Learning Goal: The power of Unix text filters
- In-Class Exercises:
- You will need these 2 files for the following questions:
/data/biocs/b/bio425/data/GBB.1con, GBB.seq- What is a genome? What does a bacterial genome typically consist of? Explain the following terms: chromosome, plasmids, and contig
- Specify the FASTA file format
- What is the size of the Borrelia burgdorferi (the Lyme disease pathogen) B31 genome in terms of
- Number of replicons: From your home directory, run:
grep -c "^>" ../../bio425/data/GBB.1con [Answer: N=22 replicons]
- Number of genes: From your home directory, run:
grep -c "^>" ../../bio425/data/GBB.seq [Answer: N=1,738 genes]
- Number of bases: First filter out FASTA headers using "grep -v" and then remove newline characters using "tr -d":
grep -v "^>" ../../bio425/data/GBB.1con | tr -d '\n' | wc -m [Answer: N=1,519,856 bases]
- Number of replicons: From your home directory, run:
- Based on the file
/data/biocs/b/bio425/data/ge-breast-cancer-cell-lines.dat, answer the following questions:- What is a transcriptome?
- How many unique genes are represented in this chip?
cut -f1 ../../bio425/data/ge-breast-cancer-cell-lines.dat | grep -vc "^Description" [Answer: N=18,900 genes]
- How many cell lines in the file?
grep "^Description" ../../bio425/data/ge-breast-cancer-cell-lines.dat| cut -f2- | wc -w [Answer: N=59 cells]
- Extract gene expression values at 3 breast cancer clinical markers: ERBB2, ESR, and PGR
grep -wP "ERBB2|ESR1|ESR2|PGR" ../../bio425/data/ge-breast-cancer-cell-lines.dat [You should see 4 rows of gene expression values]
| Assignment #1 (Final Version) |
|---|
Unix Text Filters
|
| Read Chapter 1 |
February 6. Central Dogma with PERL: Replication & Transcription
- Learning goals: String manipulations using BASH & PERL
- In-Class Exercises:
- Find out base composition (%A, %T, %C, and %G) of a DNA sequence
- In the folder
/data/biocs/b/bio425/data/GBB.1con-splitted, the whole B. burgdorferi genome has been splitted into files each of which containing a single replicon. Write a short BASH script to count how many bases in each replicon. - Write a PERL script to count GC% given a FASTA sequence.
- Run the GC-counter for each replicon using a BASH loop.
- In the folder
- DNA replication
- What is the implied directionality of a DNA sequence if none is specified?
- What is the complement strand of this DNA sequence
gatactaatgaagtatin 5' to 3' direction? - Write a PERL script to output the complementary strand of the cp9 plasmid
../../bio425/data/cp9.seqin 5' to 3' direction.
- DNA transcription
- What is the RNA transcript of this DNA sequence
gatactaatgaagtat(Make sure the answer is in 5' to 3' direction)? - Modify the DNA replication PERL script to output the RNA transcript of the cp9 plasmid
../../bio425/data/cp9.seq
- What is the RNA transcript of this DNA sequence
| Assignment #2 (Tentative; NOT final) |
|---|
|
February 13. Central Dogma with PERL: Translation & Gene Prediction
- Learning goals: DNA translation & ORF Identification
- In-Class Exercises:
- Six-frame translation
- Define ORF and reading frame. How many possible reading frames are there for a given sequence?
- How does a cell normally choose only one reading frame out of other possibilities for any given parts of a genome?
- Using a DNA genetic code table to manually translate the following sequence in all possible reading frames:
gatactaatgaagtat. Which reading frame do you think is the most likely one?
- ORF identification in a bacterial genome
- Pick a plasmid sequence from
/data/biocs/b/bio425/data/GBB.1con-splitted - Identify ORFs on the plasmid by running the program
/data/biocs/b/bio425/bin/long-orfs - Extract ORF sequences using the program
/data/biocs/b/bio425/bin/extract
- Pick a plasmid sequence from
| Assignment #2 (Tentative; NOT final) |
|---|
|
February 20 (No Class)
- Monday Schedule
February 27. Genomics with BioPerl
- (TBA)
March 6: BLAST
- (TBA)
March 13: Multiple Alignment
- (TBA)
March 20. Midterm Practicum
- (TBA)
March 27 (No Class)
Spring Break
April 3: Phylogenetics I
- (TBA)
April 10: Phylogenetics II
- (TBA)
April 17: Transcriptome with R
- (TBA)
April 24: Transcriptome with R/BioConductor
- (TBA)
May 1: Transcriptome with R/BioConductor
- (TBA)
May 8: Relational Database with SQL
- (TBA)
May 15: Review
- (TBA)
May 22: Final Project Due
Useful Links
Unix Tutorials
- A very nice UNIX tutorial (you will only need up to, and including, tutorial 4).
- FOSSWire's Unix/Linux command reference (PDF). Of use to you: "File commands", "SSH", "Searching" and "Shortcuts".
Perl Help
- Professor Stewart Weiss has taught CSCI132, a UNIX and Perl class. His slides go into much greater detail and are an invaluable resource. They can be found on his course page here.
- Perl documentation at perldoc.perl.org. Besides that, running the perldoc command before either a function (with the -f option ie, perldoc -f substr) or a perl module (ie, perldoc Bio::Seq) can get you similar results without having to leave the terminal.
Bioperl
- BioPerl's HOWTOs page.
- BioPerl-live developer documentation. (We use bioperl-live in class.)
- Yozen's tutorial on installing bioperl-live on your own Mac OS X machine. (Let me know if there are any issues!).
- A small table showing some methods for BioPerl modules with usage and return values.
SQL
- SQL Primer, written by Yozen.
R Project
- Install location and instructions for Windows
- Install location and instructions for Mac OS X
- Install R-Studio
- For users of Ubuntu/Debian:
sudo apt-get install r-base-core
Utilities
- An RSS button extension for chrome. Can add feeds to Google Reader and others.
- A similar extension which adds a "Live bookmarks"-like feature to Chrome (like Firefox's RSS bookmarks).
Other Resources
- Information Theory Primer by Thomas D. Schneider. Useful in understanding sequence logo maps.
© Weigang Qiu, Hunter College, Last Update ~~